E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 15(3); 2022 > Article
-
Case Report
Nearly Abolished Dopamine Transporter Uptake in a Patient With a Novel FBXO7 Mutation -
Eun Young Kim1
 , Seon Young Kim2, Youngduk Seo3, Chaewon Shin1,4
, Seon Young Kim2, Youngduk Seo3, Chaewon Shin1,4 -
Journal of Movement Disorders 2022;15(3):269-272.
DOI: https://doi.org/10.14802/jmd.22006
Published online: July 26, 2022
1Department of Neurology, Chungnam National University Sejong Hospital, Sejong, Korea
2Department of Laboratory Medicine, Chungnam National University Hospital, Chungnam National University, Daejeon, Korea
3Department of Nuclear Medicine, Chungnam National University Sejong Hospital, Sejong, Korea
4Department of Neurology, Chungnam National University College of Medicine, Daejeon, Korea
- Corresponding author: Chaewon Shin, MD, PhD Department of Neurology, Chungnam National University Sejong Hospital, 20 Bodeum 7-ro, Sejong 30099, Korea / Tel: +82-44-995-4751 / Fax: +82-44-995-5597 / E-mail: chw.shin@cnu.ac.kr
Copyright © 2022 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Mutations in the F-box only protein 7 (FBXO7) gene are the cause of autosomal recessive parkinsonian-pyramidal syndrome. Herein, we report a patient with a novel FBXO7 mutation with a unique clinical presentation. A 43-year-old male visited our hospital with complaints of progressing gait disturbance since a generalized tonic clonic seizure. There were no past neurological symptoms or familial disorders. Neurological examination revealed bradykinesia, masked face, stooped posture, parkinsonian gait, and postural instability. The bilateral uptake by dopamine transporters was nearly abolished, as determined by N-(3-[18F]fluoropropyl)-2β-carbon ethoxy-3β-(4-iodophenyl) nortropane positron emission tomography (18F-FP-CIT PET). Next-generation sequencing revealed a heterozygous c.1066_1069delTCTG (p.Ser356ArgfsTer56) frameshift variant and a heterozygous c.80G>A (p.Arg27His) missense variant of the FBXO7 gene. The patient’s specific clinical features, medication-refractory parkinsonism and seizures further broaden the spectrum of FBXO7 mutations. The nearly abolished dopamine transporter uptake identified by 18F-FP-CIT PET is frequently found in patients with FBXO7 mutations, which is different from the usual rostrocaudal gradient that is observed in patients with Parkinson’s disease.
- A 43-year-old male patient presented with a gait disturbance that had progressed for 10 months before the first visit. The patient grew up without any problems after having 5 to 6 afebrile seizure events before 2 years of age. He had exotropia, which developed in childhood. After graduating from university, he completed his Korean military duty service without any problems. Ten months before the visit to our hospital, he experienced a generalized tonic clonic seizure for approximately 20 minutes. Due to this seizure, he was sent to the emergency room of another hospital and was admitted for treatment of status epilepticus. Electroencephalography (EEG) did not show any abnormal findings, but he was prescribed levetiracetam 1,000 mg and lacosamide 50 mg two times per day. After discharge, the patient started to show abnormal behaviors such as stubborn personality, reduced communication, and new hobbies, such as fishing, which he had not done before. He also showed gait disturbance. At the next outpatient visit to that hospital, he was suspected to have parkinsonism and started anti-parkinsonian medications. The treatments were not effective, and he came to our outpatient clinic. The patient had a brother and sister who were normal. The parents said that the patient was normal before this seizure event but developed the problems since then. He had mild constipation but did not have other autonomic dysfunctions, hyposmia or rapid eye movement sleep behavior disorder.
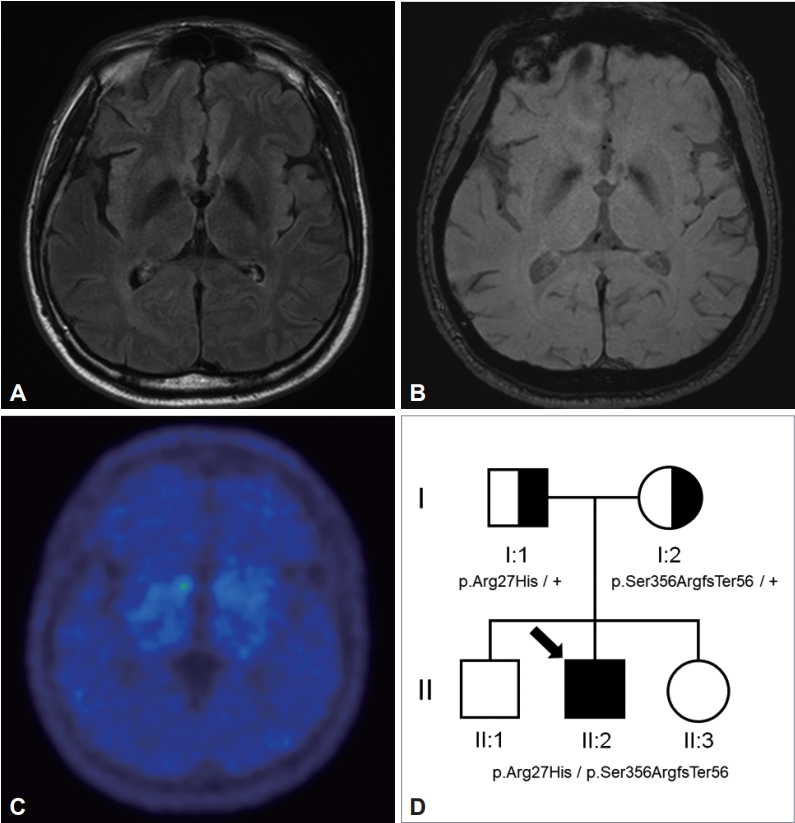
- Neurological examination revealed that the patient had exotropia, bradykinesia, masked face, stooped posture, parkinsonian gait, and postural instability (Supplementary Video 1 in the online-only Data Supplement). There was no rigidity or resting tremor, and the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale part III score was 14 points. The patient had no pyramidal signs or dystonia. The cognitive function test showed mild cognitive decline with a Mini-Mental Status Exam (MMSE) score of 28, a Montreal Cognitive Assessment score of 21, and a Clinical Dementia Rating (CDR) of 0.5 (sum-of-box 1.5). In brain magnetic resonance imaging (MRI), bilateral hypointensities in both globus palliduses were found in the susceptibility weighted imaging (SWI) sequence (Figure 1A and B). The bilateral dopamine transporter uptake was nearly eliminated, as determined by an N-(3-[18F]fluoropropyl)-2β-carbon ethoxy-3β-(4-iodophenyl) nortropane positron emission tomography (18F-FP-CIT PET) scan (Figure 1C). EEG and laboratory tests, including screening for Wilson disease, were normal.
- Next-generation sequencing was performed on 340 target genes to identify the genetic cause of his young-onset parkinsonism. These genes included ATP13A2, CHMP2B, COQ2, C9orf72, DCTN1, DNAJC6, EIF4G1, GBA, GIGYF2, GRN, HTRA2, LRRK2, MAPT, PARK2, PARK7, PDE8E, PDGFB, PINK1, PLA2G6, POLG, SLC6A3, SLC20A2, SLC30A10, SMPD1, SNCA, SOD1 SPG11, TH, UCHL1, VPS35, and FBXO7. A heterozygous c.1066_1069delTCTG (p.Ser356ArgfsTer56) frameshift variant and a heterozygous c.80G>A (p.Arg27His) missense variant of the FBXO7 gene were found. Segregation analysis showed that the FBXO7 p.Ser356ArgfsTer56 frameshift variant and the p.Arg27His missense variant were inherited from the mother and father, respectively (Figure 1D). p.Ser356ArgfsTer56 was not reported in the Asian population, and it was identified as a pathogenic variant as PM2 and PM3 in PVS1 according to the American College of Medical Genetics and Genomics guidelines. The p.Arg27His missense variant was reported previously in a Korean patient with FBXO7 mutations [2]. The minor allele frequency in the Asian population was 0.009% (gnomAD East Asian). It was predicted to be tolerated by SIFT and probably damaging by Polyphen-2 in in silico analyses. The p.Arg27His missense variant was classified as likely pathogenic (PS1, PM2, and PM3). These compound heterozygous variants are thought to be the genetic cause of the condition in this patient.
- The patient received levodopa 150 mg three times a day, pramipexole extended release form 0.75 mg and amantadine 100 mg two times a day, but there was no improvement in his symptoms. However, he showed complications such as aggressive behavior, somnolence, and impulse control disorder to gaming and shopping. Therefore, we could not increase the doses of antiparkinsonian medications. One year later, the patient’s gait abnormality had worsened, and his cognitive function deteriorated, making him more stubborn and forgetful. In the follow-up cognitive function test, there was marked dysfunction in visuospatial function, memory, and frontal/executive function on the Seoul Neuropsychological Screening Battery with MMSE 29, CDR 0.5 (sum-of-box 4.5).
CASE REPORT
- The patient was diagnosed with young-onset parkinsonism with a p.Ser356ArgfsTer56 novel frameshift and a p.Arg27His missense variant in the FBXO7 gene. Interestingly, the nearly complete loss of dopamine transporter uptake that was identified by 18F-FP-CIT PET in this case has been reported in previous case reports, and all but one of these cases harbored a FBXO7 mutation [2-5]. The only case with normal uptake showed divergent clinical characteristics, such as infantile onset, epilepsy, cerebellar degeneration, spastic paraplegia and parkinsonism [5]. This phenotypic heterogeneity might be based on differences in gene expression and brain dysfunction, which explains the normal dopamine transporter uptake.
- In patients with Parkinson’s disease (PD), the signals of dopamine receptor uptake are maintained to some extent in the striatum, such as the anterior portion of the putamen and caudate, regardless of the severity of symptoms [6]. This rostrocaudal gradient pattern is also found in genetic parkinsonism caused by LRRK2, SNCA, GBA, VPS35, and ATP1A3 mutations [7]. In contrast, patients with PINK1 and DCTN1 mutations or spinocerebellar ataxia types 2 and 3 may show a homogeneously decreased pattern similar to our case [7,8]. This pattern suggests a more widespread dopaminergic denervation and different pathophysiologic progress in patients with these genetic mutations [9]. Therefore, the loss of dopamine uptake in this case is a characteristic finding of FBXO7 mutations and several other genetic causes of parkinsonism, which is different from the usual rostrocaudal gradient that is observed in patients with idiopathic PD.
- This patient also had unique clinical characteristics that are different from those of previous reports. 1) There was a seizure event at the onset of symptoms. 2) There was a poor medication effect. 3) Brain MRI showed globus pallidus hypointensity on SWI.
- Seizures have only been reported in one case thus far [5]. FBXO7 protein is distributed widely in the frontal, temporal, and occipital cerebral cortex, hippocampus, globus pallidus, substantia nigra, and cerebellar cortex of the brain [10]. This topographic distribution can explain seizures with the severe loss of function of the FBXO7 gene. However, further studies are needed to determine its association with and pathophysiological role in seizures.
- In previous studies, the levodopa response was reported in all cases (Supplementary Table 1 in the online-only Data Supplement), except one in a low-dose trial of 100 mg/day [5]. In that case, there were hypointensities in the globus pallidus in the gradient recall echo sequence, which is similar to the present case. These results suggest that severe cases of FBXO7 mutations present with clinical findings of neurodegeneration with brain iron accumulation (NBIA) spectrum disorder, and levodopa unresponsiveness might result from postsynaptic dysfunction.
- In conclusion, this case was a patient with parkinsonism with a unique course due to a novel FBXO7 mutation. The patient’s specific clinical features, medication-refractory parkinsonism and seizures further broadened the spectrum of FBXO7 mutations. The pallidal hypointensity suggests that the disease exists in the form of an NBIA-related disorder. Finally, the nearly abolished dopamine transporter uptake that was identified by 18F-FP-CIT PET is frequently found in patients with FBXO7 mutations, and this is different from the usual rostrocaudal gradient that is observed in patients with PD.
DISCUSSION
-
Ethics Statement
Informed consent was obtained from the patient for using his case and video for publication purpose.
Notes
Supplementary Video Legends
Video 1.
Supplementary Materials
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This study was partly supported by Chungnam National University Sejong Hospital Research Fund 2021 (2021-S4-003).
-
Author Contributions
Conceptualization: Chaewon Shin. Data curation: Eun Young Kim, Chaewon Shin. Formal analysis: all authors. Funding acquisition: Chaewon Shin. Investigation: Chaewon Shin. Methodology: Eun Young Kim, Chaewon Shin. Project administration: Chaewon Shin. Resources: Seon Young Kim, Youngduk Seo, Chaewon Shin. Software: Chaewon Shin. Supervision: Chaewon Shin. Validation: Eun Young Kim, Chaewon Shin. Visualization: Eun Young Kim, Youngduk Seo, Chaewon Shin. Writing—original draft: Eun Young Kim, Chaewon Shin. Writing—review & editing: all authors.
Notes

- 1. Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA, et al. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 2008;82:1375–1384.ArticlePubMedPMC
- 2. Yoo D, Choi JH, Im JH, Kim MJ, Kim HJ, Park SS, et al. Young-onset Parkinson’s disease with impulse control disorder due to novel variants of F-Box only protein 7. J Mov Disord 2020;13:225–228.ArticlePubMedPMC
- 3. Paisán-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 2010;25:1791–1800.ArticlePubMedPMC
- 4. Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009;72:240–245.ArticlePubMed
- 5. Correa-Vela M, Lupo V, Montpeyó M, Sancho P, Marcé-Grau A, Hernández-Vara J, et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann Clin Transl Neurol 2020;7:1436–1442.ArticlePubMedPMC
- 6. Wang J, Zuo CT, Jiang YP, Guan YH, Chen ZP, Xiang JD, et al. 18F-FP-CIT PET imaging and SPM analysis of dopamine transporters in Parkinson’s disease in various Hoehn & Yahr stages. J Neurol 2007;254:185–190.ArticlePubMed
- 7. Matarazzo M, Wile D, Mackenzie M, Stoessl AJ. PET molecular imaging in familial Parkinson’s disease. Int Rev Neurobiol 2018;142:177–223.ArticlePubMed
- 8. Schöls L, Reimold M, Seidel K, Globas C, Brockmann K, Hauser TK, et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain 2015;138(Pt 11):3316–3326.ArticlePubMed
- 9. Khan NL, Valente EM, Bentivoglio AR, Wood NW, Albanese A, Brooks DJ, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol 2002;52:849–853.ArticlePubMed
- 10. Zhao T, De Graaff E, Breedveld GJ, Loda A, Severijnen LA, Wouters CH, et al. Loss of nuclear activity of the FBXO7 protein in patients with parkinsonian-pyramidal syndrome (PARK15). PLoS One 2011;6:e16983.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Imaging Procedure and Clinical Studies of [18F]FP-CIT PET
Changhwan Sung, Seung Jun Oh, Jae Seung Kim
Nuclear Medicine and Molecular Imaging.2024;[Epub] CrossRef - Study of an FBXO7 patient mutation reveals Fbxo7 and PI31 co‐regulate proteasomes and mitochondria
Sara Al Rawi, Lorna Simpson, Guðrún Agnarsdóttir, Neil Q. McDonald, Veronika Chernuha, Orly Elpeleg, Massimo Zeviani, Roger A. Barker, Ronen Spiegel, Heike Laman
The FEBS Journal.2024;[Epub] CrossRef - The characteristics of FBXO7 and its role in human diseases
Yeling Zhong, Jinyun Li, Meng Ye, Xiaofeng Jin
Gene.2023; 851: 146972. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite