E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(3); 2023 > Article
-
Review Article
Historical and More Common Nongenetic Movement Disorders From Asia -
Norlinah Mohamed Ibrahim1

 , Priya Jagota2, Pramod Kumar Pal3, Roongroj Bhidayasiri2,4, Shen-Yang Lim5,6, Yoshikazu Ugawa7, Zakiyah Aldaajani8, Beomseok Jeon9,10, Shinsuke Fujioka11, Jee-Young Lee12, Prashanth Lingappa Kukkle13,14, Huifang Shang15, Onanong Phokaewvarangkul2, Cid Diesta16, Cholpon Shambetova17, Chin-Hsien Lin18
, Priya Jagota2, Pramod Kumar Pal3, Roongroj Bhidayasiri2,4, Shen-Yang Lim5,6, Yoshikazu Ugawa7, Zakiyah Aldaajani8, Beomseok Jeon9,10, Shinsuke Fujioka11, Jee-Young Lee12, Prashanth Lingappa Kukkle13,14, Huifang Shang15, Onanong Phokaewvarangkul2, Cid Diesta16, Cholpon Shambetova17, Chin-Hsien Lin18 -
Journal of Movement Disorders 2023;16(3):248-260.
DOI: https://doi.org/10.14802/jmd.22224
Published online: June 9, 2023
1Neurology Unit, Department of Medicine, Faculty of Medicine, National University of Malaysia, Kuala Lumpur, Malaysia
2Chulalongkorn Centre of Excellence for Parkinson’s Disease and Related Disorders, Department of Medicine, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
3Department of Neurology, National Institute of Mental Health & Neurosciences, Bengaluru, Karnataka, India
4The Academy of Science, The Royal Society of Thailand, Bangkok, Thailand
5The Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
6Division of Neurology, Department of Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
7Department of Human Neurophysiology, Faculty of Medicine, Fukushima Medical University, Fukushima, Japan
8Neurology Unit, King Fahad Military Medical Complex, Dhahran, Saudi Arabia
9Department of Neurology, Seoul National University, Seoul, Korea
10Movement Disorder Center, Seoul National University Hospital, Seoul, Korea
11Department of Neurology, Fukuoka University, Faculty of Medicine, Fukuoka, Japan
12Department of Neurology, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul National University Medical College, Seoul, Korea
13Center for Parkinson’s Disease and Movement Disorders, Manipal Hospital, Bangalore, India
14Parkinson’s Disease and Movement Disorders Clinic, Bangalore, India
15Department of Neurology, Laboratory of Neurodegenerative Disorders, Rare Diseases Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
16Section of Neurology, Department of Neuroscience, Makati Medical Center, NCR, Makati, Metro Manila, Philippines
17I. K. Akhunbaev Kyrgyz State Medical Academy, Bishkek, Kyrgyzstan
18Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan
- Corresponding author: Norlinah Mohamed Ibrahim, MBBCh, FRCPE Neurology Unit, Department of Medicine, Faculty of Medicine, National University of Malaysia, Bandar Tun Razak, Kuala Lumpur 56000, Malaysia / Tel: +60-3-91456083 / E-mail: norlinah@ppukm.ukm.edu.my
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 2,062 Views
- 125 Download
- 1 Crossref
ABSTRACT
- Nongenetic movement disorders are common throughout the world. The movement disorders encountered may vary depending on the prevalence of certain disorders across various geographical regions. In this paper, we review historical and more common nongenetic movement disorders in Asia. The underlying causes of these movement disorders are diverse and include, among others, nutritional deficiencies, toxic and metabolic causes, and cultural Latah syndrome, contributed by geographical, economic, and cultural differences across Asia. The industrial revolution in Japan and Korea has led to diseases related to environmental toxin poisoning, such as Minamata disease and β-fluoroethyl acetate-associated cerebellar degeneration, respectively, while religious dietary restriction in the Indian subcontinent has led to infantile tremor syndrome related to vitamin B12 deficiency. In this review, we identify the salient features and key contributing factors in the development of these disorders.
- Asia has a total population of 4.71 billion, which represents more than 50% of the world’s population [1]. The diversity in ethnic composition and varying income tiers have directly or indirectly contributed to a number of unique movement disorders within the Asian region.
- With each disorder, there are important learning points, as the recognition of these syndromes has contributed to the knowledge of a direct cause and effect relationship and the findings of beneficial therapeutic strategies. For example, the identification of iatrogenic Creutzfeldt‒Jakob disease (CJD) has uncovered the lack of stringent irradiation procedures in the preparation of dura mater grafts. Similarly, the recognition of region-specific neurological syndromes such as amyotrophic lateral sclerosisparkinsonism dementia complex (ALS-PDC) of Kii Island, Japan, has uncovered the possibility of neurotoxin accumulation from local dietary practices, with the possibility of genetic predisposition.
- On the other hand, some disorders are purely common by virtue of a higher prevalence of predisposing medical conditions in Asia, such as diabetes mellitus (DM), certain dietary practices such as inadvertent consumption of food containing neurotoxins or exposure to industrial heavy metals. In the same vein, the unique neuropsychiatric startle syndrome Latah seems to occur only within the Southeast Asia region, among the Malay community, and is perhaps heavily influenced by their cultural practices. In this review, we summarize some of the historical and unique nongenetic movement disorders in Asia and provide the latest update on these disorders.
INTRODUCTION
- Members of the Movement Disorders in Asia Task Force of the International Parkinson and Movement Disorder Society (Asian and Oceanian Section) consisting of experts from various Asian countries, including Japan, China, Republic of Korea, Taiwan, Thailand, Malaysia, the Philippines, India, Saudi Arabia, and Kyrgyzstan, discussed and agreed upon nongenetic movement disorders that are unique and common within Asia and of historic relevance. First, all members were asked to identify movement disorders that were unique within their region, and the list of disorders was then discussed to further narrow down the disorders that are truly unique or more common in Asia. The experts finally agreed on nine disorders that fulfilled the above criteria. The syndromes reviewed in this paper are divided based on the etiology and regional prevalence within Asia. All purely infection-related movement disorders were excluded from this review.
METHODOLOGY
- Toxin- or graft-related disorders
- More than 60% of patients worldwide with dura mater graftassociated CJD (dCJD) are diagnosed in Japan [2,3]. The high incidence of dCJD in Japan is related to the remarkably frequent use of dura mater grafts in neurosurgical procedures, even in non-life-threatening conditions, such as meningioma, hemifacial spasm (HFS), and trigeminal neuralgia, compared to other countries [4]. In all cases, the operation was performed from 1975 to 1993, and the onset of dCJD was from 1985 to 2020 with an incubation period of 1–33 years (13.5 years on average) [4]. Up until September 2021, 156 patients with dCJD were identified in Japan, all of whom had received cadaveric dura mater grafts using the product Lyodura® (B. Braun, Germany) [4].
- Clinicopathologically, dCJD is classified into two distinct subtypes: plaque and nonplaque types [5-8]. The plaque type is generated by transmission of the V2 prion strain, which has valine (V) at codon 129, and type 2 protease-resistant PrP (PrPSc) to individuals with methionine (M) homozygosity (MM) at prion protein (PrP) codon 129, while the nonplaque type is caused by transmission of the M1 prion strain, which has M at codon 129, and type 1 PrPSc to individuals with MM or MV at PrP codon 129 [9,10]. The nonplaque type shows features of classical CJD, namely, rapidly progressive dementia, myoclonus, and typical electroencephalographic discharges (periodic synchronous discharges [PSDs]). On the other hand, plaque-type dCJD is atypical, characterized by relatively slow progression, lack or late occurrence of PSDs, thalamic hyperintensities on brain magnetic resonance imaging (MRI), Kuru plaques in the brain, and MM at codon 129 of the PrP gene.
- On Western blotting for PrPSc, the plaque type presents with type intermediate (type i), and transmission study with human PrP knock-in mice revealed that the plaque-type dCJD is generated by transmission of V2 prion strain, which has valine at codon 129 and type 2 PrPSc, to individuals with PrP codon 129 MM [9]. Thus, MMiK (MM at codon 129, type i PrPSc, and Kuru plaques) is a feature of plaque-type dCJD. Analysis of “sporadic CJD” from prion disease surveillance in Japan revealed that a subgroup with a history of neurosurgery without dura mater grafting had clinical features mimicking plaque-type dCJD, suggesting prion infection during operation [11].
- Experimental studies have indicated that amyloid β protein (Aβ), tau, α-synuclein, and other proteins associated with neurodegenerative diseases, such as Alzheimer’s disease or Parkinson’s disease, are transmissible via a prion-like mechanism. Analysis of these ‘proteinopathies’ in patients with dCJD showed a significant association of dura mater grafting with subpial Aβ deposition and meningeal amyloid angiopathy [12]. Aβ pathology and cerebral amyloid angiopathy (CAA) were previously reported in human growth hormone-associated CJD cases [13]. Although Aβ-type CAA commonly develops in older people showing intracerebral hemorrhage (ICH) and other cerebrovascular disorders [14], patients with early-onset, nongenetic CAA-related ICH have been recently reported with a history of neurosurgical operation with or without dura mater grafting in childhood [15,16]. A review of the literature suggests that Aβ seeds in dura mater grafts or neurosurgical instruments could cause incidental Aβ pathology, including CAA and parenchymal Aβ accumulation, and could later develop into CAA-related ICH 25 years or more after the operation [17].
- β-fluoroethyl acetate (FEA), a derivative of sodium fluoroacetate (Compound 1080, FA), is extremely toxic and has been used as a rodenticide. FEA is converted into FA, which inhibits cellular respiration, such as carbon monoxide and cyanide. FA or FEA poisoning is most often fatal because of high toxicity; thus, survivors of poisoning are extremely rare. Survivors were reported to have global encephalopathy or seizures. However, selective cerebellar atrophy as sequelae of FEA intoxication has been reported in South Korea [18,19]. Even though the production of FEA was terminated in Korea in 2005, there are 10 survivors with selective cerebellar syndrome in the literature as a historical reminder of the poor regulation of this highly toxic chemical [18]. Cerebellar dysfunction was most prominent immediately upon recovery from coma, and these patients were unable to sit, stand, or feed themselves. All patients had severe dysarthria. The neurological deficits improved over a period of months to several years but eventually plateaued with residual unsteady gait and dysarthria. Ocular motility was generally unaffected. The Mini-Mental State Examination (MMSE) scores were normal in all cases.
- Brain MRI revealed severe cerebellar atrophy, with sparing of other parts of the brain. The selective cerebellar syndrome of FEA is intriguing. FA is selective in inhibiting the metabolism and function of glial cells instead of neurons. FA is taken up selectively by glia [20]. The cerebellum has a higher mean surface density of the glial cells than other parts of the brain, which may have led to a larger FA uptake by the cerebellum [21]. The selective involvement of the cerebellum might provide a useful model for cerebellar degeneration. The tragic history of FEA emphasizes the importance of environmental regulation for public health.
- Minamata disease is a devastating neurological disease caused by mercury poisoning seen in the Minamata Bay of the Yatsushiro Sea in Kumamoto Prefecture, Japan (Figure 1) [22]. In the early 1950s, shellfish began to die, fish began to float, and seaweed stopped growing in the Minamata Bay area. One infant developed severe acute neurological symptoms such as disturbance of speech, walking, and eating in 1956. There were similar outbreaks from December 1953 to 1956, with 54 patients affected, 17 of whom had died. Subsequently, three more patients with similar symptoms presented to a hospital. This syndrome was thus reported to the Minamata Public Health Center by the hospital authorities on May 1st, 1956, which marks the official day of identification of Minamata disease. The outbreak of Minamata disease continued to spread in the coastal areas of the Yatsushiro Sea. To tackle this peculiar disorder, the Minamata City Committee for Prevention of this disease was established, and Kumamoto University established the Minamata Disease Medical Research Group. Metal poisoning was identified as the cause, with entry through the ingestion of local seafoods [23,24]. In 1959, mercury was identified as the culprit contaminant of local fish and shellfish [25]. A similar methylmercury food poisoning called ‘Niigata Minamata disease’ occurred in Niigata, Japan, between 1964 and 1965, and mercuric catalyst for acetaldehyde synthesis was again identified as the culprit for this syndrome [26]. In 1968, the government announced that Minamata disease was a toxic central nervous system disease caused by methylmercury compounds, and it was officially recognized as a pollution-related disease. After the cessation of industrial pollution by governmental regulation, no new patients were observed, even though a considerable number of the patients still had severe sequelae.
- The clinical symptoms of adult Minamata disease can be classified into two subtypes: acute and chronic poisoning. Symptoms of adult acute poisoning include visual impairment, hearing impairment, olfactory and gustatory disturbance, cerebellar ataxia, somatosensory disturbance of the distal extremities and lips, and psychiatric symptoms such as akinetic mutism, intellectual and emotional disabilities, and personality disabilities [22]. These symptoms are similar to classical methylmercury poisoning, also referred to as ‘Hunter-Russell syndrome’ [27]. In more severe cases, patients present with insanity, paralysis, coma, and eventual death within weeks of symptomatic disease onset. In chronic poisoning, adult patients typically present with glove and stocking-type paresthesia with preserved tendon reflex [27]. Cerebellar ataxia may also occur but tends to improve following cessation of exposure [28]. Visual and hearing impairment may also occur. Embryonic poisoning leads to a congenital form of the disease, which can also affect fetuses in the womb and may cause cerebral palsy. Methylmercury compounds can easily cross the blood–placental barrier, damaging the fetal brain. This is called fetal Minamata disease [29]. Patients with fetal Minamata disease have more severe intellectual disabilities, developmental disabilities, language disorders, gait disturbances, and paralysislike symptoms than adults. The diagnostic guideline for Minamata disease was proposed in 1986 [30]. The typical brain MRI findings of the disease include atrophy of the visual and postcentral cortices, cerebellar vermis, and hemisphere, and the visual cortex shows slight hypointensities on T1-weighted images [31]. There is no curative treatment, but several symptomatic therapies have been applied, including cerebellar transcranial magnetic stimulation for ataxia [32]. The mortality of the disease is estimated to be approximately 30%, and higher age is associated with a lower survival rate [33].
- Amyotrophic lateral sclerosis (ALS) was described by the Japanese 150 years ago [34]; however, endemic ALS (Muro disease) is thought to have existed in the Koza region of the Kii Peninsula (Figure 1) for more than three centuries with high incidence [35]. ALS, similar to that reported in Guam, was reported among Japanese patients in the mountainous Kii Peninsula in Japan as early as 1901, with a death rate due to motor neuron disease of 14.3 per 1,000, 23.8 times higher than the death rate due to motor neuron disease in all of Japan (0.4 per 1,000) [36]. The ALS of Kii shared similar clinical features, mode of inheritance and pathological findings to the ALS of Guam, with psychiatric manifestations such as psychosis preceding the neurological manifestations. Rapid neurological deterioration caused prominent bulbar and respiratory symptoms and resulted in death within 2 years [36], but by 1980, the disease had disappeared.
- Clinical features of the Kii ALS/Parkinsonism-dementia complex (PDC) include parkinsonism with rigidity and akinesia. Occasionally, there is tremor, bradyphrenia, abulia and amnesia, and patients can develop akinetic mutism. Over the course of the disease, patients develop clinical features that are compatible with ALS, including muscle atrophy with upper motor neuron signs and bulbar palsy. In addition, unique pigmentary retinopathy was found in one-third of the patients with Kii ALS/PDC [37]. Three clinical forms have been described in the literature: 1) classic ALS presenting with upper and lower motor neuron signs, including muscle atrophy, fasciculation, hyperreflexia and extensor plantar response; 2) PDC presenting with parkinsonism and unresponsiveness to levodopa, which may or may not be associated with frontal dementia; and 3) a combination of both ALS and PDC [38]. Neuroimaging shows marked temporal and frontal brain atrophy and a reduction in cerebral blood flow on single photon emission computed tomography. Neuropathology findings include marked loss of neurons and abundant neurofibrillary tangles, predominantly in the brainstem and temporal lobe, with PDC-type pathology showing substantia nigra and basal ganglionic degeneration [39]. A hallmark of the disease is the abundant accumulation of phosphorylated tau protein in neurons and glial cells. ALS and PDC frequently affected one individual and subsequent family members, suggesting that the primary cause may be genetic rather than environmental. Affected individuals had an “ALS-PDC” with TDP-43-positive inclusions [39]. In fact, a neuropathological study on the brains of patients with ALS-PDC of Kii identified three pathological proteinopathy subtypes, namely, tauopathy-dominant type, TDP-43 protein-dominant type and synucleinopathy-dominant type [40]. The predominant clinical features also differed between the three pathological subtypes, where atypical parkinsonism and dementia patients were prominent in the tauopathy-dominant type, ALS in the TDP-43-dominant subtype, and the synucleinopathy-dominant subtype presented with PDC [40].
- Diagnostic criteria authorized by the Japanese Society of Neurology defined the diagnosis as ‘possible’, ‘probable’ or ‘definite’, where possible refers to a current or previous inhabitant of the Muro district and surrounding areas in addition to any type 1 (ALS) or 2 (PDC); probable refers to a current or previous inhabitant of the Muro district and surrounding areas in addition to type 1 (ALS), type 2 (PDC) or type 3 (ALS + PDC) and a family history of ALS/PDC (ALS only, PDC only, or ALS and PDC); and definite fulfils the possible or probable criteria and has neuropathological findings of classic ALS neuropathology and neurofibrillary tangles or degeneration of the substantia nigra and basal ganglia or both [38]. Disease severity ranges from the patient being independent with activities of daily living to essentially being bedridden and needing full care assistance.
- As mentioned previously, the clinical and neuropathological features of Kii ALS/PDC are identical to those of Guam ALS/PDC, and both are disappearing neurodegenerative diseases. Spencer et al. [41] reported that ingestion of cycad seeds, used as a traditional medicine in New Guinea and Guam, as well as on the Kii Peninsula, is postulated as a cause of the disease, with the decline in incidence hypothetically linked to changes in lifestyle, including cessation of the use of cycad seeds in these communities. On the other hand, a targeted genomic sequencing study on patients with ALS-PDC of Guam identified homozygous PINK1 p.L347P, heterozygous DCTN1 p.T54I, FUS p. P431L, and HTT (42 CAG repeats) as pathogenic mutations. The authors concluded that these genetic mutations could contribute to the genetic and clinical heterogeneity of the disease within these communities [42].
- Cultural syndrome
- Latah syndrome is a culturally stereotyped startle behavior in response to variable sudden and unexpected external stimuli, resulting in echolalia, coprolalia and echopraxia, occurring almost exclusively among women of the Malay ethnic groups in Malaysia (formerly Malaya) and Indonesia [43-46]. It was first described in the 19th century as a mental malady or disease of the Malays [44].
- One of the earliest scientific papers described a series of patients with Latah syndrome triggered by sudden and unexpected tactile, auditory, or verbal stimuli. Two phenotypes of Latah syndrome were described depending on the predominant manifestations: the mimetic type, in which involuntary and often unwilling mimicry of actions was the main symptom, and the paroxysmal type, in which coprolalia (or repetitive verbalization) was the main symptom [44]. The mimetic type resulted in an unwilling yet unstoppable mimicry of the actions of someone else, almost like in a hypnotic state. Following this, the sufferer would feel remorseful, apologetic, and often angry toward the provocateur. The paroxysmal type was shorter lasting, resulting in coprolalia (which was stereotyped) and suggestible behaviors that were variable, such as the utterance of obscene words repetitively (such as in the vocal tics of Tourette’s syndrome) to more socially unacceptable behaviors, such as undressing in public [44]. These initial clinical descriptions of Latah syndrome vary somewhat from the more recent understanding of the syndrome, which is now believed to be a hyperekplexic syndrome. A more comprehensive evaluation of Latah syndrome in 1978 summarized the syndrome as a shock or startle syndrome, often occurring in postmenopausal women of Malay ethnicity (although not exclusively) and leading to vulgar (often sexual) utterances and latah behavior [45].
- A systematic evaluation of 12 patients with Latah syndrome with detailed neurological evaluations and neurophysiological testing was supportive of Latah syndrome being a neuropsychiatric startle syndrome [47]. A recent review highlighted similarities in the phenomenology of movements to motor tics, but unlike tics, the movements in Latah syndrome were stimulus-induced and not preceded by premonitory symptoms [46]. The bizarre and paroxysmal nature of the movements with suggestibility favored a psychogenic cause. However, some patients had exaggerated nose-tap head retraction reflex, a hallmark of hereditary hyperekplexia. There was an increase in early motor startle reflex, as seen in patients with hereditary hyperekplexia (GRA1 and GLYT2) syndrome, although not to the same extent. Increased early motor startle reflex was also reported in patients with anxiety disorders, further supporting Latah syndrome as a neuropsychiatric startle syndrome disorder.
- Neurometabolic syndromes
- Infantile tremor syndrome (ITS) is characterized by tremors, anemia, skin depigmentation and developmental delay occurring in children between the ages of 5 months and 3 years [48,49]. The syndrome was first reported in India in 1957 by Dikshit et al. [50] as ‘nutritional dystrophy and anemia’, and later, the term ‘infantile tremor syndrome’ was coined by Bajpai et al. [51] in 1965. It is most commonly reported from the Indian subcontinent, Southeast Asian countries and Africa, but the exact incidence is unknown. In India, it accounts for 0.2%–2% of pediatric hospital admissions [52]. Mothers of infants with ITS are mostly vegetarians, and ITS occurs exclusively in breast-fed infants.
- The etiology of ITS is unknown. Nutritional deficiency, viral infections and degenerative hypotheses have been postulated [48]. Vitamin B12 deficiency is most often implicated [53], while other possible etiologies include protein–energy malnutrition/undernutrition, mineral deficiency (e.g., Mg and Zn) and enzyme defects (tyrosine).
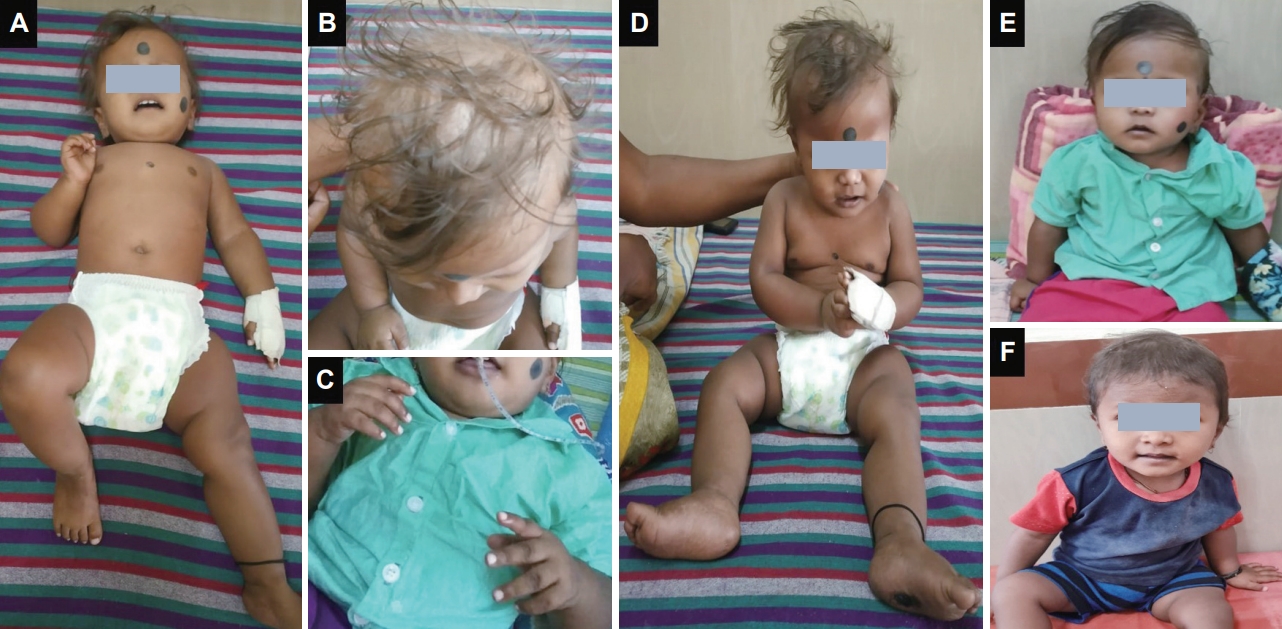
- Three stages of ITS have been described: pretremor, tremor and posttremor. The children are usually born without any perinatal complications and have normal neurodevelopment until the first 4–6 months. Developmental slowing then sets in, followed by somnolence and lethargy. In the pretremor phase, there is neuromotor regression and sometimes a tremulous voice. The patients usually have pallor, knuckle pigmentation, a reticulate pattern of hyperpigmentation over the trunk and limbs, and sparse, depigmented hair (Figure 2). The infant looks dull but plump with preserved subcutaneous fat. Anemia, angular cheilitis, stomatitis, glossitis, rickets, scurvy, and edema may be present. Mild hepatomegaly with or without splenomegaly is frequently reported. Congestive cardiac failure secondary to anemia may occur. Seizures are uncommon but may occur at any time during the course of the disease.
- The tremor phase is heralded by sudden onset of tremors involving various parts of the body, mainly the hands and feet [54]. Tremors are coarse and jerky (myoclonus-like), usually start focally in one upper limb, and rapidly spread to the whole body (generalized pattern) (Supplementary Videos 1 and 2 in the online-only Data Supplement). Sudden onset of generalized tremors is not uncommon. Even when generalized, tremors are asymmetric and multifocal. The involvement of the facial, labial, lingual, and laryngeal musculature is also common. Laryngeal involvement renders a tremulous character to the vocalization/cry, which resembles the bleating of a goat. Tremors may be intermittent initially, brought about by crying, excitement, or physical stimuli. Later tremors become constant throughout the day but disappear during sleep. In severe cases, tremors may persist even during sleep, although at reduced intensity. Other movement disorders, such as choreoathetosis, myoclonus, dystonia and orolingual movements, can also occur, although they are underrecognized [55,56]. Although the limb muscles usually feel flabby, there is rigidity on passive movements with retained or exaggerated tendon reflexes [54].
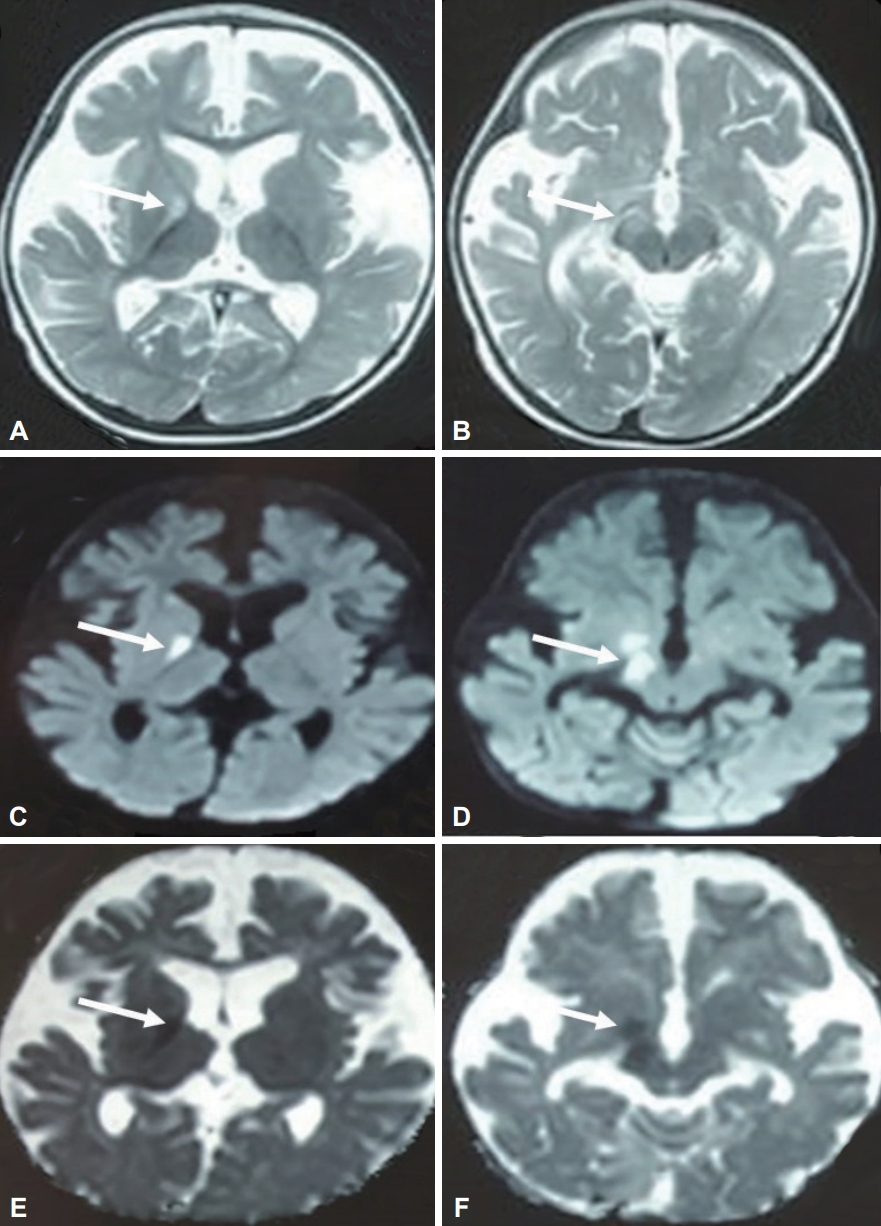
- Neuroimaging features include diffuse nonspecific cerebral atrophy, cerebellar atrophy, reduced thickness of the corpus callosum, delayed myelination/hypomyelination, subdural effusion, prominence of the Sylvian sulcus, dilatation of the ventricular system, T2 hyperintensities in the globus pallidum and bright signals on diffusion-weighted image (DWI) suggestive of restricted diffusion (Figure 3). Cerebral atrophy and globus pallidus changes have been reported to be reversible at follow-up [56,57].
- Management of ITS is largely empirical and includes vitamin B12, folic acid, iron, calcium, zinc, and magnesium supplementation and a high-protein diet [58]. There is gradual recovery over weeks to months irrespective of the treatment. Vitamin B12 alone was shown to result in dramatic improvement. General activity and responsiveness improve within 48–72 hours, and lost milestones begin to return. Tremors disappear within a week in most infants and by 3–4 weeks in all infants. Tremors can be treated with phenobarbitone, chlorpromazine, carbamazepine and propranolol [59,60]. In a recent study, tremor favorably responded to propranolol [52]. The mean duration of treatment needed to completely control the tremor was 35.3 days, ranging from 7 to 75 days. Hematological recovery occurs within 5–7 days, skin pigmentation resolves in 2–4 weeks, and hair changes improve over several months to years. Although most infants ultimately recover, neurodevelopmental deficits, especially those related to cognitive and language skills, often persist for a long time. Mortality in ITS is very low and is usually due to intercurrent infections.
- Kernicterus refers to the clinical features of chronic bilirubin encephalopathy secondary to unconjugated bilirubin levels close to or higher than 20 mg/d. Untreated, it can lead to extrapyramidal syndrome, sensorineural hearing loss, and dental enamel dysplasia.
- The prevalence of Kernicterus is unknown. A Swedish study reported that hazardous hyperbilirubinemia in near-term or term newborns was associated with disabling brain damage in 13 per one million births [61]. In Middle East countries, kernicterus is commonly due to underlying glucose-6-phosphate dehydrogenase deficiency [62], the latter being more prevalent in this region due to a high degree of marital consanguinity.
- The long-term sequelae of kernicterus encompass 1) abnormal motor control, abnormal movements, and a decrease in muscle tone, which at a later stage is replaced by spasticity and rigidity; 2) symmetrical auditory processing disturbance with or without hearing loss; 3) oculomotor impairments involving upward vertical gaze leading to “the setting sun sign”; and 4) dysplasia of the enamel of deciduous teeth. Additional features include intelligence and emotional disturbances, epilepsy, speech difficulties and, rarely, myelopathy [63].
- The involvement of the globus pallidus and subthalamic nucleus in kernicterus leads to dyskinesia and choreoathetotic movements [63]. In severe cases, children may develop athetotic or dyskinetic forms of cerebral palsy. Mild kernicterus may present with only dystonia and mild motor milestone delays. There may be difficulty in ambulation due to choreoathetotic movements. The severe form is characterized by marked dystonia and athetosis, which interfere with controlled voluntary movements such as ambulation, speech and self-feeding. Lack of coordination or generalized clumsiness and hypotonia may occur. Swallowing and eating can be difficult due to spasms of the pharyngeal muscles and involuntary movements of the tongue.
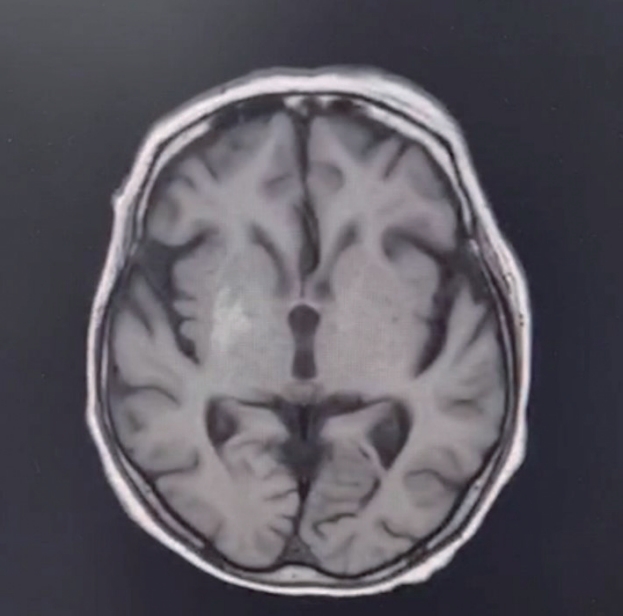
- The brain MRI findings are characteristic, with hyperintense signals in the bilateral globus pallidus on T1-weighted sequences and bilateral, symmetrical hyperintense signals on T2-weighted and fluid attenuated inversion recovery (FLAIR) sequences in the globus pallidus and subthalamic nuclei, which are considered the hallmark radiological features of the disease (Figure 4). Pathological findings include yellow discoloration of the basal ganglia, particularly of the globus pallidus and subthalamic nucleus, indicating that the urobilinogen had crossed the blood-brain barrier [64].
- Perinatal hyperbilirubinemia needs to be treated aggressively to prevent the chronic development of kernicterus and its devastating sequelae. In addition to supportive management of neurological sequelae, patients may require medications and surgical treatment depending on the severity and type of movement disorder. Although kernicterus has become less common presently in most parts of the world due to advanced neonatal care, some cases are still seen in less privileged areas of the world where delayed access to health care plays a major role [63].
- Hemichorea-hemiballism (HCHB) associated with hyperglycemia, also known as diabetic striatopathy, is frequently reported among Asians [65]. It is relatively rare, with an estimated prevalence of 1 in 100,000 worldwide [66]. Historically, HCHB-hyperglycemia syndrome was first reported in 1960 by Bedwell SF in a black patient who presented with recurrent and reversible transient episodes of hemiballism, which correlated with blood glucose elevations [67]. Subsequently, similar cases were reported in the 1980s, with recovery following the correction of hyperglycemia [68-70]. The first Asian case was reported in 1990 in a patient who presented with reversible chorea associated with nonketotic hyperglycemia and sepsis [71]. A recent systematic review of 176 cases of diabetic striatopathy from 1992 until 2018 identified that 71.6% of the cases reported were from Asia, followed by Europe and America [72].
- The clinical presentation is typically of an acute or subacute onset hemichorea or hemiballism (most commonly in an arm-leg distribution), although rarely bilateral chorea, monochorea [65], alternating hemichorea [70], or generalized chorea may also occur (Supplementary Videos 3 and 4 in the online-only Data Supplement) [66,72]. The onset is typically within days of the hyperglycemic episode, although rarely the onset may be delayed for up to a month [65,72]. In 17% of patients, HCHB may be the first presentation of DM [72]. The mainstay of treatment is rapid correction of hyperglycemia with insulin, with or without anti-chorea medications such as haloperidol, clonazepam, or other dopamine receptor antagonists. In most cases, HCHB resolves fully, although recurrence was reported in 14% of patients [72]. While elderly patients with type 2 DM are most predisposed, there are recent reports of this syndrome also occurring in pediatric patients with type 1 DM [73]. Females have a higher predilection than males at a ratio of 1.7:1 [72]. Apart from being more frequent among Asians, there are no differential clinical features that distinguish HCHB among Asians compared to non-Asian ethnic groups.
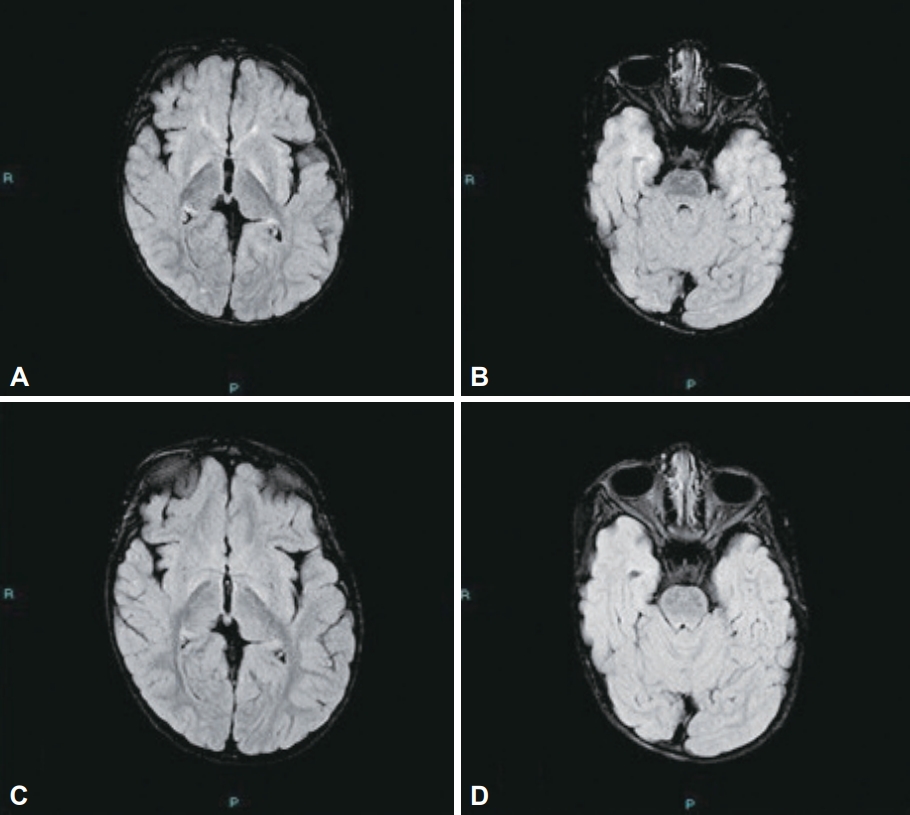
- Computed tomography (CT) scans show characteristic unilateral striatal hyperdensity lesions contralateral to the HCHB (Figure 5), with corresponding hyperintensity on T1-weighted MRI and typically hypointense, sometimes rarely isointense T2-weighted MRI changes [72,74]. The absence of perilesional edema and the sparing of the internal capsule distinguishes this syndrome from hemorrhage [75]. Bilateral striatal abnormalities on both CT and MRI have been reported [72]. The putamen is most commonly involved, followed by the caudate and globus pallidus [65,74]. Extrastriatal involvement affecting the contralateral thalamus, midbrain, pontine and cerebellar peduncle regions [76] and the contralateral medial temporal, anterior temporal and insula regions resulting in temporal lobe atrophy has rarely been reported [77]. Other MRI findings include high signal intensity on DWI with low apparent diffusion coefficient (ADC) values on ADC map [78], high signal on T2/FLAIR and hypointensity on SWI [72]. MRI has a better yield in the detection of striatal abnormalities compared to CT, as the latter may be negative in a small percentage of patients [75]. Brain imaging abnormalities are typically reversible within days, mirroring the improvement in hyperglycemia, but may take up to 6 months in some cases [72]. Radiological resolution occurs later than clinical resolution, with normalization of CT changes occurring earlier than MRI [74].
- The underlying pathogenesis is still not fully understood. The presence of restricted diffusion and the persistence of radiological abnormalities months after the correction of hyperglycemia indicate that there may be mechanisms other than hyperviscosity-related ischemia alone [72]. Pathological findings include petechial hemorrhage, calcium deposition, reactive astrocytosis and abundant gemistocytes [72,76]. Reduced glucose metabolism in the affected regions on fluorodeoxyglucose-positron emission tomography (FDG-PET) and 18-F-FDG-PET [79] and a low NAA/Cr ratio on magnetic resonance spectroscopy suggest neuronal dysfunction or metabolic failure [74].
- This HCHB-hyperglycemia syndrome is now well recognized in Asia as one of the reversible causes of metabolic movement disorder. Although there are now increasing reports of this syndrome in other populations, this syndrome will remain more prevalent in Asia given the higher prevalence of DM here. In addition, Asian patients have been shown to have higher postprandial blood glucose levels and greater blood glucose excursions compared to the Caucasian population, which could also contribute to the higher prevalence among Asians [80]. The possibility of a genetic predisposition among Asians should also be explored in the future.
- HFS is a peripherally induced movement disorder that most commonly develops in mid to late adulthood. It typically begins in the orbicularis oculi before spreading to the other muscles of facial expression and usually occurs unilaterally. HFS is socially embarrassing, significantly impacts quality of life, and at times can be disabling.
- To date, there are no direct comparative population-based epidemiological data to confirm the long-held anecdotal observation that HFS is more common in Asians. Epidemiologic studies performed in the United States and Norway reported prevalences of approximately 10 per 100,000 population [81], but such data are not available from Asian countries. Nevertheless, a clinic-based study conducted by Wu et al. [82] in Houston, Texas, USA, found that Asian patients disproportionately accounted for 10.6% of all HFS patients under follow-up, whereas Caucasians (with an approximately 10-fold larger population in the Houston Metropolitan Area) accounted for 61.4%. The authors extrapolated these figures to provide prevalence figures of 12.6 and 7.2 per 100,000 for the Asian and Caucasian communities, respectively. Study limitations included a unicentric design and modest sample size (total n = 132 patients with HFS). Additional indirect support for a high prevalence of HFS in Asians is the relatively large sample sizes of reported HFS databases from Asian countries such as Thailand, China, India, Japan, and the Philippines [83-88].
- HFS is commonly attributed to posterior circulation arterial loops compressing the seventh cranial nerve as it exits the pons, leading to ephaptic transmission (i.e., lateral spread of excitation from one demyelinated neuron to another adjacent neuron) and involuntary facial muscle contractions. A relatively smaller posterior fossa in the Asian skull, with crowding of the cranial nerves and vascular structures—making neurovascular compression more likely to occur—has been proposed as a mechanism contributing to the increased frequency of HFS in Asians. Indeed, a study from Singapore using high-resolution three-dimensional MRI volumetric analysis demonstrated that patients with HFS had a smaller mean volume of the posterior fossa cerebrospinal fluid (CSF) space than that of controls [89]. Female sex was also associated with a smaller posterior fossa CSF volume, potentially explaining the female preponderance in HFS [90]. A Japanese study using brain computerized tomography scans similarly showed a narrower cerebellopontine angle cistern in patients with HFS than in controls [91].
- Hypertension has been associated with HFS, but whether this is causal (e.g., high blood pressure contributing to ectatic or tortuous blood vessels [92]) or merely correlative (e.g., vessel loops causing both HFS and compression of the ventrolateral medulla with resultant hypertension) is unclear [93].
- To our knowledge, there have been no studies of potential genetic susceptibility factors in sporadic HFS. Interestingly, a large kindred of English origin with Charcot-Marie-Tooth disease type 1B was reported to exhibit a high frequency (8/27 = 29.6%) of HFS, with or without trigeminal neuralgia (with 30% having bilateral involvement, vs. approximately 3% in sporadic cases) [94]. This occurrence was posited to be possibly due to factors intrinsic to the nerve (e.g., nerve hypertrophy increasing the risk of neural compression or other factors reducing the threshold for aberrant discharges) [94]. There is a need for further epidemiological, genetic, imaging and anatomic studies to understand the prevalence differences and pathophysiology of this common condition.
RESULTS
Dura mater graft-related iatrogenic CJD in Japan
β-Fluoroethyl acetate-associated cerebellar degeneration in Korea
Minamata disease in Japan
Amyotrophic lateral sclerosis of the Kii Peninsula
Latah syndrome in Malaysia, Indonesia and Thailand
Infantile tremor syndrome in India
Kernicterus in the middle east
Hemichorea-hemiballism associated with hyperglycemia
Hemifacial spasm
- In this review, we have summarized the historical and common nongenetic diseases from Asia, which are of clinical relevance and historical importance, and highlight key messages and important lessons that can be derived from these disorders. While some of the diseases described have disappeared completely, disorders such as ITS and kernicterus are still prevalent within certain communities and need to be recognized and treated appropriately. Historical and devastating toxin-related diseases such as Minamata disease in Japan and FEA poisoning emphasize the importance of the regulation of toxic products and wastes in light of urbanization and industrialization. The predilection of Latah syndrome in certain ethnic groups indicates the possibility of culturally driven behavioral mimicry or common genetic predisposition. Similarly, the reduced prevalence of ALS-PDC in Kii reveals that environmental toxins could play an important role in other neurodegenerative diseases as well. For more common disorders such as HFS and HCHB hyperglycemia syndrome, this review supports the fact that these disorders may indeed be more prevalent among Asians.
DISCUSSION AND CONCLUSION
Supplementary Material
Video 1.
Video 2.
Video 3.
Video 4.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None
-
Author contributions
Conceptualization: all authors. Methodology: all authors. Writing—original draft: Norlinah Mohamed Ibrahim, Priya Jagota, Shen-Yang Lim, Pramod Kumar Pal, Jee-Young Lee, Prashanth Lingappa Kukkle, Shinsuke Fujioka, Huifang Shang, Onanong Phokaewvarangkul, Roongroj Bhidayasiri, Yoshikazu Ugawa, Zakiyah Aldaajani, Chin-Hsien Lin. Writing—review & editing: all authors.
Notes
- We would like to acknowledge Prof. Vykunta Raju KN (Department of Paediatric Neurology, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India) for the videos and figures provided for infantile tremor syndrome.
Acknowledgments



- 1. United Nations Department of Economic and Social Affair. World Population Prospects 2019 Highlights. [Internet]. New York: United Nations; c2019 [accessed on 2022 Sep 22]. Available at: https://population.un.org/wpp/publications/files/wpp2019_highlights.pdf.
- 2. Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, et al. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis 2012;18:901–907.ArticlePubMedPMC
- 3. Ae R, Hamaguchi T, Nakamura Y, Yamada M, Tsukamoto T, Mizusawa H, et al. Update: dura mater graft-associated Creutzfeldt-Jakob disease - Japan, 1975-2017. MMWR Morb Mortal Wkly Rep 2018;67:274–278.ArticlePubMedPMC
- 4. Hamaguchi T, Sakai K, Noguchi-Shinohara M, Nozaki I, Takumi I, Sanjo N, et al. Insight into the frequent occurrence of dura mater graft-associated Creutzfeldt-Jakob disease in Japan. J Neurol Neurosurg Psychiatry 2013;84:1171–1175.ArticlePubMed
- 5. Noguchi-Shinohara M, Hamaguchi T, Kitamoto T, Sato T, Nakamura Y, Mizusawa H, et al. Clinical features and diagnosis of dura mater graft associated Creutzfeldt Jakob disease. Neurology 2007;69:360–367.ArticlePubMed
- 6. Yamada M, Noguchi-Shinohara M, Hamaguchi T, Nozaki I, Kitamoto T, Sato T, et al. Dura mater graft-associated Creutzfeldt-Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology 2009;29:609–618.ArticlePubMed
- 7. Sakai K, Hamaguchi T, Noguchi-Shinohara M, Nozaki I, Takumi I, Sanjo N, et al. Graft-related disease progression in dura mater graft-associated Creutzfeldt-Jakob disease: a cross-sectional study. BMJ Open 2013;3:e003400. ArticlePubMedPMC
- 8. Sakai K, Hamaguchi T, Sanjo N, Murai H, Iwasaki Y, Hamano T, et al. Diffusion-weighted magnetic resonance imaging in dura mater graft-associated Creutzfeldt-Jakob disease. J Neurol Sci 2020;418:117094.ArticlePubMed
- 9. Kobayashi A, Asano M, Mohri S, Kitamoto T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 2007;282:30022–30028.ArticlePubMed
- 10. Kobayashi A, Parchi P, Yamada M, Mohri S, Kitamoto T. Neuropathological and biochemical criteria to identify acquired Creutzfeldt-Jakob disease among presumed sporadic cases. Neuropathology 2016;36:305–310.ArticlePubMed
- 11. Hamaguchi T, Sakai K, Kobayashi A, Kitamoto T, Ae R, Nakamura Y, et al. Characterization of sporadic Creutzfeldt-Jakob disease and history of neurosurgery to identify potential iatrogenic cases. Emerg Infect Dis 2020;26:1140–1146.ArticlePubMedPMC
- 12. Hamaguchi T, Taniguchi Y, Sakai K, Kitamoto T, Takao M, Murayama S, et al. Significant association of cadaveric dura mater grafting with subpial Aβ deposition and meningeal amyloid angiopathy. Acta Neuropathol 2016;132:313–315.ArticlePubMedPDF
- 13. Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015;525:247–250.ArticlePubMedPDF
- 14. Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke 2015;17:17–30.ArticlePubMedPMC
- 15. Hamaguchi T, Komatsu J, Sakai K, Noguchi-Shinohara M, Aoki S, Ikeuchi T, et al. Cerebral hemorrhagic stroke associated with cerebral amyloid angiopathy in young adults about 3 decades after neurosurgeries in their infancy. J Neurol Sci 2019;399:3–5.ArticlePubMed
- 16. Jaunmuktane Z, Quaegebeur A, Taipa R, Viana-Baptista M, Barbosa R, Koriath C, et al. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol 2018;135:671–679.ArticlePubMedPMCPDF
- 17. Yamada M, Hamaguchi T, Sakai K. Acquired cerebral amyloid angiopathy: an emerging concept. Prog Mol Biol Transl Sci 2019;168:85–95.ArticlePubMed
- 18. Kim JM, Jeon BS. Survivors from beta-fluoroethyl acetate poisoning show a selective cerebellar syndrome. J Neurol Neurosurg Psychiatry 2009;80:528–532.PubMed
- 19. Jin JH, Lee ES, Choi JY, Kim JS. Isolated cerebellar atrophy due to rodenticide (β-fluoroethyl acetate) intoxication. J Neurol Sci 2017;373:208–209.ArticlePubMed
- 20. Szerb JC, Issekutz B. Increase in the stimulation-induced overflow of glutamate by fluoroacetate, a selective inhibitor of the glial tricarboxylic cycle. Brain Res 1987;410:116–120.ArticlePubMed
- 21. Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 1998;18:8751–8757.ArticlePubMedPMC
- 22. Ekino S, Susa M, Ninomiya T, Imamura K, Kitamura T. Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J Neurol Sci 2007;262:131–144.ArticlePubMed
- 23. Yorifuji T. Lessons from an early-stage epidemiological study of Minamata disease. J Epidemiol 2020;30:12–14.ArticlePubMedPMC
- 24. Kitamura S, Miyata C, Tomita M, Date S, Kojima T, Minamoto H, et al. A central nervous system disease of unknown cause that occurred in the Minamata region: results of an epidemiological study. J Epidemiol 2020;30:3–11.ArticlePubMedPMC
- 25. Takeuchi T, Kambara T, Morikawa N, Matsumoto H, Shiraishi Y, Ito H. Pathologic observations of the Minamata disease. Acta Pathol Jpn 1959;9(Suppl):769–783.Article
- 26. Shimohata T, Hirota K, Takahashi H, Nishizawa M. [Clinical aspects of the Niigata Minamata disease]. Brain Nerve 2015;67:31–38.Japanese. PubMed
- 27. Hunter D, Russell DS. Focal cerebellar and cerebellar atrophy in a human subject due to organic mercury compounds. J Neurol Neurosurg Psychiatry 1954;17:235–241.PubMedPMC
- 28. Ninomiya T, Imamura K, Kuwahata M, Kindaichi M, Susa M, Ekino S. Reappraisal of somatosensory disorders in methylmercury poisoning. Neurotoxicol Teratol 2005;27:643–653.ArticlePubMed
- 29. Matsumoto H, Koya G, Takeuchi T. Fetal Minamata disease. A neuropathological study of two cases of intrauterine intoxication by a methyl mercury compound. J Neuropathol Exp Neurol 1965;24:563–574.PubMed
- 30. Eto K. Pathology of Minamata disease. Toxicol Pathol 1997;25:614–623.ArticlePubMedPDF
- 31. Korogi Y, Takahashi M, Okajima T, Eto K. MR findings of Minamata disease--organic mercury poisoning. J Magn Reson Imaging 1998;8:308–316.ArticlePubMed
- 32. Nakamura M, Bekki M, Miura Y, Itatani M, Jie LX. Cerebellar transcranial magnetic stimulation improves ataxia in Minamata disease. Case Rep Neurol 2019;11:167–172.ArticlePubMedPMCPDF
- 33. Tamashiro H, Arakaki M, Akagi H, Futatsuka M, Roht LH. Mortality and survival for Minamata disease. Int J Epidemiol 1985;14:582–588.ArticlePubMed
- 34. Miura K. On amyotrophic lateral sclerosis. J Neurol (Shinkeigaku Zasshi, Tokyo) 1902;191:1–15.
- 35. Uebayashi Y, Yase Y, Tanaka H, Shimada Y, Toyokura Y. Prognosis of motor neuron disease in Japan. Neuroepidemiology 1983;2(3-4):243–256.ArticlePDF
- 36. Yase Y. VII. Neurologic disease in the Western Pacific Islands, with a report on the focus of amyotrophic lateral sclerosis found in the Kii peninsula, Japan. Am J Trop Med Hyg 1970;19:155–166.ArticlePubMed
- 37. Kokubo Y, Ito K, Kuzuhara S. Ophthalmomyiasis-like pigmentary retinopathy in ALS/PDC in the Kii peninsula of Japan. Neurology 2003;60:1725–1726.ArticlePubMed
- 38. Kokubo Y. [Diagnostic criteria for amyotrophic lateral sclerosis/parkinsonism-dementia complex in the Kii peninsula, Japan]. Brain Nerve 2015;67:961–966.Japanese. PubMed
- 39. Kuzuhara S, Kokubo Y. Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update. Mov Disord 2005;20 Suppl 12:S108–S113.ArticlePubMed
- 40. Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii peninsula: a multiple proteinopathy? Neuropathology 2018;38:98–107.ArticlePubMedPDF
- 41. Spencer PS. Parkinsonism and motor neuron disorders: lessons from Western Pacific ALS/PDC. J Neurol Sci 2022;433:120021.ArticlePubMed
- 42. Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 2015;77:458–468.ArticlePubMed
- 43. Bartholomew RE. Disease, disorder, or deception? Latah as habit in a Malay extended family. J Nerv Ment Dis 1994;182:331–338.discussion 339-341. ArticlePubMed
- 44. Ellis WG. Latah. a mental Madady of the Malays. J Ment Sci 1897;43:32–40.Article
- 45. Kenny MG. Latah: the symbolism of a putative mental disorder. Cult Med Psychiatry 1978;2:209–231.ArticlePubMedPDF
- 46. Lim TT, Tan K, Eow GB, Bhidayasiri R. A South East Asian perspective of neuropsychiatric startle syndromes of latah. Parkinsonism Relat Disord 2022;95:138–142.ArticlePubMed
- 47. Bakker MJ, van Dijk JG, Pramono A, Sutarni S, Tijssen MA. Latah: an Indonesian startle syndrome. Mov Disord 2013;28:370–379.ArticlePubMedPDF
- 48. Kalra V. Infantile tremor syndrome. In: Ghai OP, Paul VK, Bagga A, editors. GHAI Essential Pediatrics. 7th ed. New Delhi: CBS Publishers and Distributors Pvt Ltd; 2009:558–559.
- 49. Sharda B, Bhandari B. Infantile tremor syndrome. Indian Pediatr 1987;24:415–421.PubMed
- 50. Dikshit AK. Nutritional dystrophy and anaemia. Ind J Child Health 1957;6:132–139.
- 51. Bajpai PC, Tandon PN, Sharma NL, Misra PK. Infantile tremor syndrome. Acta Neurol Scand 1965;41:473–486.ArticlePubMed
- 52. Gowda VK, Kolli V, Benakappa A, Srinivasan VM, Shivappa SK, Benakappa N. Case series of infantile tremor syndrome in tertiary care paediatric centre from southern India. J Trop Pediatr 2018;64:284–288.ArticlePubMed
- 53. Goraya JS, Kaur S. Infantile tremor syndrome: a review and critical appraisal of its etiology. J Pediatr Neurosci 2016;11:298–304.ArticlePubMedPMC
- 54. Bajpai PC, Misra PK, Tandon PN. Further observations on infantile tremor syndrome. Indian Pediatr 1968;5:297–307.PubMed
- 55. Sharawat IK, Kasinathan A, Sankhyan N. Infantile tremor syndrome: response to B12 therapy. J Pediatr 2018;196:323–323.e1.ArticlePubMed
- 56. Kesavan S, Dhawan S, Saini L, Attri SV, Vyas S, Sankhyan N. Reversible basal ganglia changes and metabolic crisis in infantile tremor syndrome. Indian J Pediatr 2020;87:464–465.ArticlePubMedPDF
- 57. Gupta R, Rawat AK, Singh P, Gupta J, Pathak A. Infantile tremor syndrome: current perspectives. Res Rep Trop Med 2019;10:103–108.PubMedPMC
- 58. Ghai OP, Gupta P. Infantile tremor syndrome. In: Paul VK, Bagga A, editors. Ghai Essential Pediatrics. 8th ed. New Delhi: CBS Publishers and Distributors Pvt Ltd; 2013:580–581.
- 59. Ratageri VH, Shepur TA, Patil MM, Hakeem MA. Scurvy in infantile tremor syndrome. Indian J Pediatr 2005;72:883–884.ArticlePubMedPDF
- 60. Murali MV, Sharma PP, Koul PB, Gupta P. Carbamazepine therapy for infantile tremor syndrome. Indian Pediatr 1993;30:72–74.PubMed
- 61. Alkén J, Håkansson S, Ekéus C, Gustafson P, Norman M. Rates of extreme neonatal hyperbilirubinemia and kernicterus in children and adherence to national guidelines for screening, diagnosis, and treatment in Sweden. JAMA Netw Open 2019;2:e190858. ArticlePubMedPMC
- 62. Nair PA, Al Khusaiby SM. Kernicterus and G6PD deficiency--a case series from Oman. J Trop Pediatr 2003;49:74–77.ArticlePubMed
- 63. Ali ZAE. The sequelae of kernicterus [abstract]. Mov Disord 2019;34(suppl 2):Abstract No. 498. Available at: https://www.mdsabstracts.org/abstract/the-sequelae-of-kernicterus/.
- 64. Hamza A. Kernicterus. Autops Case Rep 2019;9:e2018057. ArticlePubMedPMC
- 65. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a meta-analysis of 53 cases including four present cases. J Neurol Sci 2002;200(1-2):57–62.PubMed
- 66. Ondo WG. Hyperglycemic nonketotic states and other metabolic imbalances. Handb Clin Neurol 2011;100:287–291.ArticlePubMed
- 67. Bedwell SF. Some observations on hemiballismus. Neurology 1960;10:619–622.ArticlePubMed
- 68. Rector WG Jr, Herlong HF, Moses H III. Nonketotic hyperglycemia appearing as choreoathetosis or ballism. Arch Intern Med 1982;142:154–155.ArticlePubMed
- 69. Totoritis M, Cornish D, Thompson F. Nonketotic hyperglycemia. Arch Intern Med 1982;142:1405.Article
- 70. Sanfield JA, Finkel J, Lewis S, Rosen SG. Alternating choreoathetosis associated with uncontrolled diabetes mellitus and basal ganglia calcification. Diabetes Care 1986;9:100–101.ArticlePDF
- 71. Lim TO, Ngah BC. Diabetic non-ketotic hyperglycaemia presenting as chorea--a case report. Med J Malaysia 1990;45:260–262.PubMed
- 72. Chua CB, Sun CK, Hsu CW, Tai YC, Liang CY, Tsai IT. “Diabetic striatopathy”: clinical presentations, controversy, pathogenesis, treatments, and outcomes. Sci Rep 2020;10:1594.ArticlePubMedPMCPDF
- 73. Rai S, Kaul V, Singh S, Kaur S, Thenmurugan P. Diabetic striatopathy: a new challenge in type 1 pediatric diabetic patients. Oman Med J 2022;37:e332. ArticlePubMedPMC
- 74. Johari B, Hanafiah M, Shahizon AMM, Koshy M. Unilateral striatal CT and MRI changes secondary to non-ketotic hyperglycaemia. BMJ Case Rep 2014;2014:bcr2014204053.ArticlePubMedPMC
- 75. Lin JJ, Lin GY, Shih C, Shen WC. Presentation of striatal hyperintensity on T1-weighted MRI in patients with hemiballism-hemichorea caused by non-ketotic hyperglycemia: report of seven new cases and a review of literature. J Neurol 2001;248:750–755.ArticlePubMedPDF
- 76. Zhang Y, Parikh A. Clinical and neuroimaging features in a patient with non-ketotic hyperglycemia. Neurol Int 2020;12:130–135.ArticlePubMedPMC
- 77. Kammeyer RM, Orjuela KD. Rapidly progressive dementia and temporal lobe atrophy in a case of nonketotic hyperglycemic hemichorea. Neurohospitalist 2020;10:229–233.ArticlePubMedPMCPDF
- 78. Chu K, Kang DW, Kim DE, Park SH, Roh JK. Diffusion-weighted and gradient echo magnetic resonance findings of hemichorea-hemiballismus associated with diabetic hyperglycemia: a hyperviscosity syndrome? Arch Neurol 2002;59:448–452.ArticlePubMed
- 79. Moon S, Kim HE, Oh TJ, Ahn CH, Choi SH, Jang HC. Hyperglycemic hemichorea: a case series. Ann Geriatr Med Res 2021;25:222–225.ArticlePubMedPMCPDF
- 80. Zhang XM, Li PF, Hou JN, Ji LN. Blood glucose profiles in East Asian and Caucasian injection-naive patients with type 2 diabetes inadequately controlled on oral medication: a pooled analysis. Diabetes Metab Res Rev 2018;34:e3062. ArticlePubMedPMCPDF
- 81. Nilsen B, Le KD, Dietrichs E. Prevalence of hemifacial spasm in Oslo, Norway. Neurology 2004;63:1532–1533.ArticlePubMed
- 82. Wu Y, Davidson AL, Pan T, Jankovic J. Asian over-representation among patients with hemifacial spasm compared to patients with cranial-cervical dystonia. J Neurol Sci 2010;298(1-2):61–63.ArticlePubMed
- 83. Poungvarin N, Devahastin V, Viriyavejakul A. Treatment of various movement disorders with botulinum A toxin injection: an experience of 900 patients. J Med Assoc Thai 1995;78:281–288.PubMed
- 84. Yuan Y, Wang Y, Zhang SX, Zhang L, Li R, Guo J. Microvascular decompression in patients with hemifacial spasm: report of 1200 cases. Chin Med J (Engl) 2005;118:833–836.PubMed
- 85. Wang L, Hu X, Dong H, Wang W, Huang Y, Jin L, et al. Clinical features and treatment status of hemifacial spasm in China. Chin Med J (Engl) 2014;127:845–849.ArticlePubMed
- 86. Batla A, Goyal C, Shukla G, Goyal V, Srivastava A, Behari M. Hemifacial spasm: clinical characteristics of 321 Indian patients. J Neurol 2012;259:1561–1565.ArticlePubMedPDF
- 87. Mizobuchi Y, Nagahiro S, Kondo A, Arita K, Date I, Fujii Y, et al. Prospective, multicenter clinical study of microvascular decompression for hemifacial spasm. Neurosurgery 2021;88:846–854.ArticlePubMedPDF
- 88. Yu JRT, Jamora RDG, Silverio EL, Bautista JMP, Luspian KJL, Tiongson RM, et al. Spectrum of movement disorders in two movement disorders centers in the Philippines. Acta Neurol Taiwan 2021;30:94–101.
- 89. Chan LL, Ng KM, Fook-Chong S, Lo YL, Tan EK. Three-dimensional MR volumetric analysis of the posterior fossa CSF space in hemifacial spasm. Neurology 2009;73:1054–1057.ArticlePubMedPMC
- 90. Zhang H, Yin X, Ouyang Z, Chen J, Zhou S, Zhang C, et al. A prospective study of freezing of gait with early Parkinson disease in Chinese patients. Medicine (Baltimore) 2016;95:e4056. ArticlePubMedPMC
- 91. Kamiguchi H, Ohira T, Ochiai M, Kawase T. Computed tomographic analysis of hemifacial spasm: narrowing of the posterior fossa as a possible facilitating factor for neurovascular compression. J Neurol Neurosurg Psychiatry 1997;62:532–534.ArticlePubMedPMC
- 92. Weiss D, Cavinato C, Gray A, Ramachandra AB, Avril S, Humphrey JD, et al. Mechanics-driven mechanobiological mechanisms of arterial tortuosity. Sci Adv 2020;6:eabd3574. ArticlePubMedPMC
- 93. Leong JL, Li HH, Chan LL, Tan EK. Revisiting the link between hypertension and hemifacial spasm. Sci Rep 2016;6:21082.ArticlePubMedPMCPDF
- 94. Caress JB, Lewis JA, Pinyan CW, Lawson VH. A charcot-marie-tooth type 1B kindred associated with hemifacial spasm and trigeminal neuralgia. Muscle Nerve 2019;60:62–66.ArticlePubMedPMCPDF
REFERENCES
Figure & Data
References
Citations
- Diabetic striatopathy and other acute onset de novo movement disorders in hyperglycemia
Subhankar Chatterjee, Ritwik Ghosh, Payel Biswas, Shambaditya Das, Samya Sengupta, Souvik Dubey, Biman Kanti Ray, Alak Pandit, Julián Benito-León, Rana Bhattacharjee
Diabetes & Metabolic Syndrome: Clinical Research & Reviews.2024; 18(3): 102997. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite