E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(3); 2023 > Article
-
Letter to the editor
Clinical and Genetic Features of Huntington’s Disease Patients From Republic of Serbia: A Single-Center Experience -
Nikola Kresojević1,2

 , Ivana Perović2, Iva Stanković1,2, Aleksandra Tomić1,2, Milica Jecˇmenica Lukic´1,2, Vladana Marković1,2, Tanja Stojković1,2, Gorana Mandić1,2, Milena Janković1, Ana Marjanović1, Marija Branković1, Ivana Novaković2, Igor Petrović1,2, Nataša Dragašević1,2, Elka Stefanova1,2, Marina Svetel1,2, Vladimir Kostić1,2
, Ivana Perović2, Iva Stanković1,2, Aleksandra Tomić1,2, Milica Jecˇmenica Lukic´1,2, Vladana Marković1,2, Tanja Stojković1,2, Gorana Mandić1,2, Milena Janković1, Ana Marjanović1, Marija Branković1, Ivana Novaković2, Igor Petrović1,2, Nataša Dragašević1,2, Elka Stefanova1,2, Marina Svetel1,2, Vladimir Kostić1,2 -
Journal of Movement Disorders 2023;16(3):333-335.
DOI: https://doi.org/10.14802/jmd.23028
Published online: June 9, 2023
1Neurology Clinic, University Clinical Center of Serbia, Belgrade, Republic of Serbia
2Faculty of Medicine, University of Belgrade, Belgrade, Republic of Serbia
- Corresponding author: Nikola Kresojevic´ , MD, PhD Neurology Clinic, University Clinical Center of Serbia, Dr Subotic´ a 6, Belgrade 11000, Republic of Serbia / Tel: +381-112685596 / Fax: +381-112684577 / E-mail: nikola_kresojevic@yahoo.com
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,166 Views
- 70 Download
- Dear Editor,
- Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an expansion of CAG trinucleotide repeats in the HTT gene, which encodes the huntingtin protein [1]. The normal number of CAG repeats in the HTT gene is between 10 and 26; if there are 27–35 repeats, HD will not occur, but due to meiotic instability, the number of repeats may increase, and the disease may manifest in offspring. A total of 36–39 repeats represent a gray zone with reduced penetrance, where the symptoms occur only in some individuals. Finally, if there are more than 40 CAG repeats, the penetrance is complete, and the disease will certainly manifest [1].
- Here, we report the clinical features of patients diagnosed with HD at Neurology Clinic, University Clinical Center of Serbia. A previous publication regarding the survival of HD patients in Republic of Serbia was the result of a follow-up study (from 1982 to 2004) in movement disorders department [2]. However, the data presented in the current article were collected from an electronic database, which was introduced in Neurology Clinic, University Clinical Center of Serbia in 2003; therefore, some patients may have been included in both studies.
- A total of 125 patients were found to have sufficient clinical data, including 53 males (42.4%) and 72 females (57.6%). The average age at disease onset (AAO) in our cohort was 45.4 ± 13.3 years (range 13–82 years), similar to the median age of onset of 43 years found in a larger cohort of 1,766 HD patients [3]. Juvenile onset is generally rare, occurring in approximately 5% of HD patients with a high number of trinucleotide CAG repeats (over 60) [4]. In our cohort, only three patients with 64, 71, and 81 CAG repeats had juvenile onset. The small number of juvenile onset patients in our sample may result from referral bias of the tertiary center for adult patients.
- A total of 16 (14%) patients had a negative family history of HD, a result similar to the rate of 10% reported in the literature and attributed to the anticipation phenomenon in individuals with an intermediate number of CAG repeats (27–35) [1,5].
- In our sample, the most common initial symptom was chorea in 56% of patients, which occurred at a mean age of 47.4 ± 12.5 years (range 14–82 years); 32% had cognitive-psychiatric symptoms (at 41.5 ± 14.0 years, range 13–70 years), and 8.8% had mixed symptoms (at 49.0 ± 14.0 years, range 23–69 years). A similar distribution of initial symptoms was found in a larger multicenter cohort including 1,766 patients, of whom 48% had motor symptoms, 28% had cognitive-psychiatric symptoms (19.6% with psychiatric symptoms and 8.4% with cognitive symptoms), and 13.2% had mixed symptoms [3]. We did not find a difference in the number of CAG repeats when comparing the group of patients with psychiatric symptoms at the disease onset and the group of patients with motor symptoms at the disease onset (p = 0.829). In patients with motor symptoms at the disease onset, cognitive-psychiatric symptoms appeared 3.5 ± 3.5 years later (range 0–12 years), and in those with initial cognitive-psychiatric symptoms, chorea occurred after 3.8 ± 4.8 years (range 0–24 years), reflecting the progressive nature of HD.
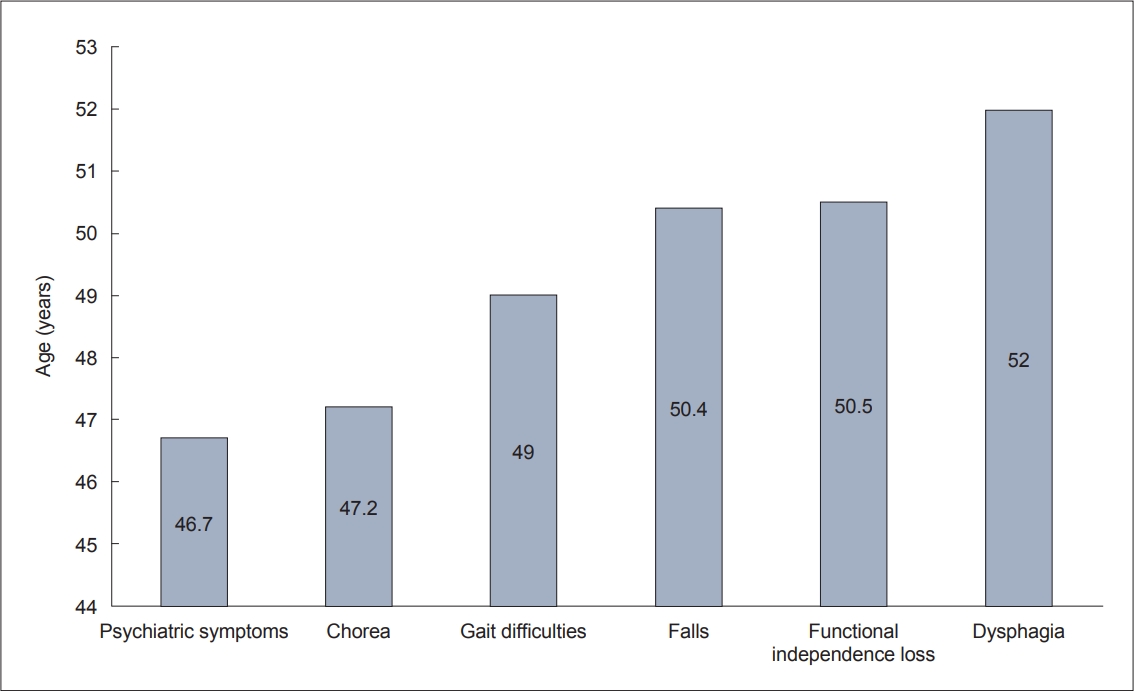
- We found that difficulty walking and falls occurred around the age of 50 years (49.0 ± 13.2 and 50.4 ± 13.8 years, respectively). Falls may result in serious injury, and according to the results of Busse et al. [6], only 20.8% of patients did not report falls after one year of follow-up. In our cohort, eight (6.4%) patients suffered significant injuries from falls, including hip or rib fractures and arm or leg injuries. Dysphagia is a late manifestation of HD that may lead to aspiration pneumonia and acute respiratory distress syndrome [7]. In our sample, dysphagia occurred in 41.2% of patients and manifested at a mean age of 52.0 ± 13.1 years (range 17–75 years) (Figure 1).
- Motor and cognitive symptoms and depression are significant predictors of the functional loss of abilities in HD patients [8]. All HD patients will eventually develop some type of psychiatric disorder; Sellers et al. [9] reported that 100% of HD patients have at least one psychiatric disorder, most often apathy and irritability. Chorea has less impact on patients’ functionality than bradykinesia, rigidity, and coordination and gait problems, which occur in later stages of the disease [10,11]. Typically, the loss of work abilities and capacities occurs in the early disease stage, while in the advanced disease stage, 24-hour care from another person is often necessary [12]. In our cohort, the loss of functional independence occurred at a mean age of 50.5 ± 13.4 years (range 15–85 years), at approximately the same time as the onset of gait disorders and falls. Death can result from complications due to falls, malnutrition, aspiration, pneumonia, or suicide [10,11,13].
- The average number of CAG repeats in the pathological allele was 45 ± 6 (range 40–81, median 44) in our sample. We found a strong negative correlation between the number of CAG repeats and the AAO (Pearson’s correlation coefficient r = -0.745, p < 0.001), the onset of the first psychiatric symptoms (r = -0.726, p < 0.001), chorea (r = -0.754, p < 0.001), difficulty walking (r = -0.759, p < 0.001), dysphagia (r = -0.728, p < 0.001) and the loss of functional independence (r = -0.785, p < 0.001). The number of CAG repeats affects the AAO and, to a lesser extent, the degree of disease progression [11]. The higher the number of CAG repeats, the earlier the AAO and the greater the progression. The length of CAG repeats can explain 50%–70% of the variance in the AAO and can be influenced by the sex of the parent who transmits the mutated gene; meiotic instability of an intermediate number of CAG repeats exists in paternal transmission [1,11].
- In conclusion, the analysis of the clinical data of HD patients available at Neurology Clinic, University Clinical Center of Serbia showed typical HD symptoms with disease onset in adulthood, relatively rapid full clinical expression of cardinal symptoms, and loss of patient functionality. The number of CAG trinucleotide repeats had a strong negative correlation with the age of disease onset but also with other clinical markers of disease progression.
-
Ethics Statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the ethical board of the University Clinical Center of Serbia (IRB #: NK75/2020) and with the 2013 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all the patients included in the study.
-
Conflicts of Interest
Nikola Kresojević, Milica Ječmenica Lukić, Igor Petrović, Nataša Dragašević, Marina Svetel, have received a speaker honorarium from Salveo. The funder has no role to publish this article. All remaining authors have declared no conflicts of interest.
-
Funding Statement
This study was supported by the Ministry of Education and Science of the Republic of Serbia (Grant #175090 to VK).
-
Author contributions
Conceptualisation: Nikola Kresojević, Ivana Perović. Data curation: Ivana Perović, Aleksandra Tomić, Milica Ječmenica Lukić, Tanja Stojković, Gorana Mandić. Formal analysis: Iva Stanković, Vladana Marković. Funding aquisition: Vladimir Kostić. Investigation: Milena Janković, Ana Marjanović, Marija Brankovič, Ivana Novaković. Methodology: Igor Petrović, Nataša Dragašević, Elka Stefanova. Supervision: Marina Svetel. Writing—original draft: Nikola Kresojević, Ivana Perović. Writing—review & editing: Vladimir Kostić.
Notes
- 1. Myers RH. Huntington’s disease genetics. NeuroRx 2004;1:255–262.ArticlePubMedPMC
- 2. Pekmezovic T, Svetel M, Maric J, Dujmovic-Basuroski I, Dragasevic N, Keckarevic M, et al. Survival of huntington’s disease patients in Serbia: longer survival in female patients. Eur J Epidemiol 2007;22:523–526.ArticlePubMedPDF
- 3. Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, et al. Observing Huntington’s disease: the European Huntington’s Disease Network’s REGISTRY. PLoS Curr 2010;2:RRN1184.ArticlePubMedPMC
- 4. Quarrell OW, Nance MA, Nopoulos P, Paulsen JS, Smith JA, Squitieri F. Managing juvenile Huntington’s disease. Neurodegener Dis Manag 2013;3:10.2217/nmt.13.18. Article
- 5. Baig SS, Strong M, Quarrell OW. The global prevalence of Huntington’s disease: a systematic review and discussion. Neurodegener Dis Manag 2016;6:331–343.ArticlePubMed
- 6. Busse ME, Wiles CM, Rosser AE. Mobility and falls in people with Huntington’s disease. J Neurol Neurosurg Psychiatry 2009;80:88–90.ArticlePubMed
- 7. Heemskerk AW, Roos RA. Dysphagia in Huntington’s disease: a review. Dysphagia 2011;26:62–66.ArticlePubMedPDF
- 8. Beglinger LJ, O’Rourke JJ, Wang C, Langbehn DR, Duff K, Paulsen JS, et al. Earliest functional declines in Huntington disease. Psychiatry Res 2010;178:414–418.ArticlePubMedPMC
- 9. Sellers J, Ridner SH, Claassen DO. A systematic review of neuropsychiatric symptoms and functional capacity in Huntington’s disease. J Neuropsychiatry Clin Neurosci 2020;32:109–124.ArticlePubMed
- 10. Walker FO. Huntington’s disease. Lancet 2007;369:218–228.ArticlePubMed
- 11. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10:204–216.ArticlePubMedPDF
- 12. McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol 2018;25:24–34.ArticlePubMedPDF
- 13. Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010;5:40.ArticlePubMedPMCPDF
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite