E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 3(2); 2010 > Article
-
Review Article
X-Linked Dystonia Parkinsonism: Clinical Phenotype, Genetics and Therapeutics - Raymond L. Rosalesa,b
-
Journal of Movement Disorders 2010;3(2):32-38.
DOI: https://doi.org/10.14802/jmd.10009
Published online: October 30, 2010
aDepartment of Neurology and Psychiatry, University of Santo Tomas, Manila, Philippines
bCNS-Center for Neurodiagnostic and Therapeutic Services, Metropolitan Medical Center, Manila, Philippines
- Corresponding author: Raymond L. Rosales, MD, PhD, CNS-Center for Neurodiagnostic and Therapeutic Services, Metropolitan Medical Center, 3001 MAB, University of Santo Tomas Hospital Sampaloc, Manila 1008, Philippines, Tel +63-2-252-7080, Fax +63-2-254-7432, E-mail rrosales@info.com.ph
Copyright © 2010 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 42,602 Views
- 348 Download
- 27 Crossref
ABSTRACT
- The clinical phenotype of X-Linked Dystonia Parkinsonism (XDP) is typically one that involves a Filipino adult male whose ancestry is mostly traced in the Philippine island of Panay. Dystonia usually starts focally in the lower limbs or oromandibular regions, then spreads to become generalized eventually. Parkinsonism sets in later into the disease and usually in combination with dystonia. /DYT3/ and /TAF1/ are the two genes associated with XDP. An SVA retrotransposon insertion in an intron of /TAF1/ may reduce neuron-specific expression of the /TAF1/ isoform in the caudate nucleus, and subsequently interfere with the transcription of many neuronal genes. Polypharmacy with oral benzodiazepines, anticholinergic agents and muscle relaxants leaves much to be desired in terms of efficacy. The medications to date that may appear beneficial, especially in disabling dystonias, are zolpidem, muscle afferent block with lidocaine-ethanol and botulinum toxin type A. Despite the few cases undergoing deep brain stimulation, this functional surgery has shown the greatest promise in XDP. An illustrative case of XDP in a family depicts the variable course of illness, including a bout of “status dystonicus,” challenges in therapy, reckoning with the social impact of the disease, and eventual patient demise. Indeed, there remains some gaps in understanding some phenomenological, genetic and treatment aspects of XDP, the areas upon which future research directions may be worthwhile.
- From the latest account (as of February 2010) of Lee and cohorts,2 there are now 505 cases from 253 families in the registry of the XDP study. They estimate that the present prevalence rate for the entire Philippines is 0.31 per 100,000 population. For the island of Panay, their estimated prevalence is 5.74 per 100,000, with the province of Capiz having the highest rate (23.66 per 100,000 estimate), followed by the provinces of Aklan, Iloilo, Antique and Guimaras, in that order. Whilst these figures are estimates, additional XDP cases have been found in the other islands of the three main regions of the Philippines (e.g., Bicol, Mindoro, Masbate, Romblom and Palawan provinces of the Luzon region; Negros, Samar and Leyte provinces of the Visayas region; and Davao and Cotabato provinces of the Mindanao region, among other places). People are quite known to migrate outside the country due to petitions and employment needs, hence it is not unusual to find these affected Filipinos with XDP in countries like the USA, Canada, Germany, UK and Japan. Endeavors to further trace the ancestries of these other XDP patients to the island of Panay were possible in some, but certainly not all of them. Hence, this gap in epidemiology is a piece of a puzzle that may be needed in our understanding of XDP in terms of ancestry, migration and the possible “gene-environment interaction.”
Prevalence
- Maintaining a national registry, Lee and coworkers of the XDP study group3–5 gave detailed clinical accounts, and with the recent updated data consisting of 505 cases from 253 families, they were quick to conclude that the clinical picture and natural course of XDP only slightly changed.2 Age of onset is usually 39–40 years (range 12–64 years), mean duration of illness is 16 years, and mean age at death is 55.6 years. To date, five affected females have been identified, giving a male/female gender ratio of 100 : 1. All the female cases have affected brothers and most likely with carrier mothers as evidenced by the presence of affected relatives in both parents.2,6 Independent clinical XDP reviews from elsewhere have likewise alluded to parallel findings.7–10
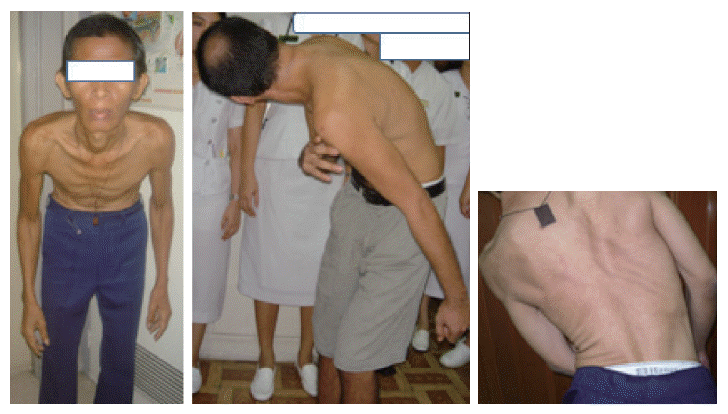
- Compared to parkinsonian traits as onset symptoms, more than 90% of XDP cases initially present with focal dystonia, when evident, parkinsonism typically manifests with slow and periodic resting tremor, bradykinesia, micrographia, hypomimia and shuffling gait. The more prevalent focal dystonia present in the following descending order of occurrence: Lower extremities (31%) > craniofacial area (28%) > neck & shoulder (23%) > upper extremities (14%) and trunk (2.6%).2 The following are the common clinical manifestations of the dytonias per region, roughly arranged in order of frequency: Big toe dorsiflexion (“striatal toe”), foot extension/flexion/inversion, toe fanning and knee extension/flexion (-lower extremities); jaw opening/closing/deviation, tongue protrusion/rolling/retraction, blepharospasm, twitching of the face, mouth pursing, snout-like movements of lips and adductor laryngeal dysphonia (-craniofacial region); rotational (torticollis), laterocollis, retrocollis, anterocollis, (or a combination thereof), with and without shoulder elevation (-neck and shoulder regions); wrist extension/ flexion, writer’s cramp involving fingers, and elbow extension/flexion (-upper extremities); spinal flexion/extension/ lateral deviation (or a combination thereof) and flexion at the pelvis or camptocormia (-truncal region)(Figure 1).
- Regardless of the first site of regional involvement, the initial focal dystonia in XDP spreads in 97% of the time and generalizes within 5 years in 84%,2 some of whom may be segmental in the interim. With disease progression, the predominant phenotype is one of nearly constant, severe dystonic movements usually affecting the axial areas, with prominent oromandibular involvement such as jaw opening and tongue protrusion. “Geste antagonistique” may initially abolish the oromandibular, lingual, cervical and truncal dystonias, but eventually fail as the dystonia progresses in time and in regional involvement.
- Typically between the 2nd to the 7th year of illness, the dystonic phase predominate, however, as early as the second year, parkinsonian traits may appear. As the illness reaches the 7th to the 10th year, both dystonia and parkinsonism now co-exist; labeled as the combined dystonia- parkinsonian phase. By the 15th year of illness, the predominant picture is one of parkinsonism manifesting as tremors, bradykinesia, masked facies, hypomimia and drooling; labeled as the parkinsonian phase. About 68% of the patients on the average are predominantly dystonic throughout and only 18.6% are predominantly parkinsonian. The small number of cases who reach the parkinsonian phase may be due to the shortened life span (mean of 51 yrs) of XDP cases.2 Aerodigestive dysfunction leading to malnutrition and aspiration pneumonia are the usual causes of demise in XDP, however, the growing number of suicides, whether independent, or within families, has become a new challenge to treating physicians. Important personal and social factors to consider are family history of suicide, depression, anxiety, disfigurement, sleep impairment, “catastrophizing” (thus impending severe disability and death), “stigmatization” (thus social withdrawal and isolation), family separation/disintegration and loss of employment (i.e., men being in the most productive ages of their working careers).
- In recognition of the aforementioned phases, the XDP study group proposed an XDP Disease Staging that categorizes extent and severity of the condition by grading patients based on three parameters: 1) The clinical presentation (whether the movement disorder involves a part or several parts of the body and whether the patient manifests dystonic or parkinsonian features or both); 2) The degree of functional impairment in performance of activities of daily living, and 3) the need for caregiver assistance or being independent. Thus, for Stage I: focal dystonia or one parkinsonian component, with no functional impairment and is independent; for Stage II: segmental dystonia or one parkinsonian component, with no functional impairment and is independent; for Stage III: Multifocal dystonia or any dystonia plus parkinsonian component(s), with mild functional impairment and still independent; for Stage IV: Generalized dystonia or any dystonia plus parkinsonian component(s), with moderate to severe functional impairment and is dependent; and for Stage V: Any combination of dystonia and parkinsonism components, bedridden and highly dependent. In case any of the three components is of a more advanced stage, the final overall stage of the illness will be the stage where the most severe component belongs.2
- Regarding manifesting females with XDP, an initial report alluded to a milder course and that they do not present with dystonia, or if they do, the dystonia is usually focal, non-progressive, and non-disabling.6 A scrutiny on the clinical profile of the affected females in the XDP registry indicate that their clinical presentation do not significantly vary from their male counterparts. In fact, an affected female in the family we have carefully followed up had severe manifestations of progressive dystonia, including parkinsonism, as were her two affected brothers.
Clinical phenotype
- Imaging and neuropathology help us shed light into the interplay of pathophysiology and genetics in XDP. Cranial MRI in XDP shows hyperintense putaminal rim during both dystonic and parkinsonian phases, and atrophy of the caudate head or putamen during the parkinsonian phase.11 Neuropathology confirms an atrophy of the caudate nucleus and putamen, with mild to severe neuronal loss and gliosis.12 During the dystonic phase of XDP, the neostriatum shows involvement of striosomes, but with matrix sparing, whilst during the parkinsonian phase, matrix involvement occurs as well. During the dystonic phase, the loss of striosomal inhibitory projections lead to disinhibition of nigral dopaminergic neurons, likely resulting to a hyperkinetic state; whilst in the parkinsonian phase, severe and critical reduction of matrix-based projection may result in extranigral parkinsonism.13
- It is believed that XDP originated ancestrally in the Philippines, particularly in the Panay Islands through a founder mutation some 50 meiotic generations (−1,000 years) ago.5,10 Two genes are associated with XDP: /DYT3/and /TAF1/(TATA binding protein-associated factor-1).14,15 Gene sequencing of the XDP critical region in Xq13.1 revealed an SVA retrotransposon insertion in an intron 32 of /TAF1/. This phenomenon may effectively reduce neuron-specific expression of the/TAF1/isoform in the caudate nucleus, and subsequently interfere with the transcription of many neuronal genes. The caveat is that analysis of /TAF1/ is not easy, as the gene contains many alternative splicing isoforms and is a member of a larger complex (hence it was previously regarded as multiple transcript system), transcription factor IID (TFIID). TFIID is a DNA-binding complex required for RNA polymerase II and plays a role in the initiation of transcription as it binds to the TATA element. SVA retrotransposon insertions are thought to be active in the human genome and to alter the expression level of adjacent genes that cause diseases. Furthermore, the SVA retrotransposon has a high GC content (approximately 70%) and a large number of CpG sites (more than 150 sites) in its nucleotide sequence, so that it is frequently hypermethylated in its insertion site (May Christine Malicdan, personal communication). The entire pathomechanism of this disease still remains enigmatic. It has been proposed that differential neuronal death might cause an imbalance in activity of striatosomal and matrix-based pathways thus giving rise to abnormal movement. Whilst in XDP, the decreased expression of the neuron-specific TA14–391 isoform and probably other /TAF1/ isoforms, may result in transcriptional dysregulation of many neuronal genes, including that which encodes dopamine receptor, /DRD2/,15 a robust evidence is awaited to demonstrate this matter.
- At the moment, the function of /TAF1/ remains to be elaborated, and how it leads to disease can only be answered by indepth studies using cultured cell models and animal models. Likewise, though the groups of Makino15 and Nolte14 have found “disease specific” nucleotide variants in /DYT3/, a robust functional evidence remains wanting. Moreover, /DYT3/ is still a putative gene at the moment and the protein product has not been purified. At this time, what can be said is that patients carry Disease specific Single nucleotide Changes (DSC) in /DYT3/. Nolte and colleagues14 have also found a 48bp deletion in exon 4 of /TAF1/, but this has not been replicated by other groups. Although it is possible to identify DSCs haplotype and SVA retrotransposon insertion, which may be used as a routine molecular genetic diagnosis of XDP cases, blood /TAF1/ and gene /CXCR3/ expression profiling may not reliable surrogate markers for this disorder.16 Finally, in regard to the affected females in XDP, the implication of a possible codominant disorder or extreme X-inactivation mosaicism is possible (albeit the fact that one symptomatic woman was homozygous for the mutation).5,6,17,18
Pathophysiology and genetics
- Oral medications
- Published data using various oral medications for XDP are very scarce. Of the reported 28 cases from the then known as “Torsion Dystonia of Panay,” 20 cases have been tried on of anti-parkinsonian drugs, anti-histaminics, and phenothiazines. Whilst no mention to which specific symptoms and to which extent of improvement occurred, one case each reported partial response to levodopa/carbidopa and to haloperidol.1 Virtually, oral medications in the form of benzodiazepines (e.g., clonazepam, diazepam), anticholinergic agents (e.g., biperiden, trihexyphenidyl), antipsychotic agents (e.g., haloperidol) and antihistamines (e.g., diphenhydramine), including anti-parkinsonian medications (e.g., levodopa/carbidopa), have failed to show convincing efficacy in XDP.4 Milacemide (top dose of 4,800 mg/day) given in one XDP case with severe freezing of gait (case 8 from the reported 10 cases with various neurodegenerative conditions), failed to show benefit and despite having on board diazepam (30 mg/ day), trihexyphenidyl (30 mg/ day), and levodopa (300 mg/day).19
- Perhaps to date the only drug showing potential benefit in XDP is zolpidem. Zolpidem, an imidazopyridine, is a nonbenzodiazepine hypnotic with a selective agonist effect for GABA type A receptors. The binding sites for zolpidem are most abundant in the output structures of the basal ganglia (i.e., globus pallidus pars interna or GPi and substantia nigra pars reticularis).20 In movement disorders, zolpidem improves akinesia, dystonia and dyskinesia in advanced Parkinson’s disease,21 improves parkinsonism in progressive supranuclear palsy,22 and oppose dopaminergic-induced dyskinesia.23 A study of zolpidem in three cases of XDP indicated its usefulness once dystonia becomes multifocal or generalized, and where polypharmacy offers only partial relief.24 In predominantly phasic type of generalized dystonic movements, zolpidem may induce nearly 100% improvement of dystonia for a few hours. The clinical effect of zolpidem may last six to eight hours per 10 mg dose in the first few weeks. Subsequently, the effect becomes progressively shorter, decreasing to two to three hours.10 Addition of zolpidem may induce cross-tolerance in instances where adverse events of drugs may be an issue in view of initial use of benzodiazepines,25 Clinical trials on zolpidem are underway involving larger XDP cohorts, including the awaited final stages of levodopa/cabidopa trial.
- Chemodenervation
- Chemodenervation in XDP has been reported applying muscle afferent block (MAB) and botulinum toxin type A (BoNT-A), especially during the focal, multifocal and segmental stages of the dystonia. The aims of the therapy are geared toward the disabling dystonia and associated pain. MAB was previously performed in an open-label study of 30 XDP cases injecting 0.5% lidocaine and 95% ethanol, in a volume ratio of 10 : 1.26 Electromyography (EMG)-guided injections of MAB were administered to the dystonic tongue, jaw, neck, trunk, upper and lower limb muscles. Depending upon injected muscle size, the dose ranged from 5–20 mL MAB, care being taken that the total lidocaine dose per patient, per day, did not exceed 1 mL/ kg body weight. Employing predetermined outcome assessments, there was an overall improvement in the EMG turns parameters and a moderate improvement in the subjective scales. Mean onset of MAB effect was 27.3 minutes, and the mean duration of effect lasted 7.9 days. Transient and non-serious adverse events were seen in 11/30 cases that included dizziness and injection site pain, bleeding and erythema. Blocking muscle afferents with diluted lidocaine (MAB) to effectively abolish cervical dystonia (CD) and writer’s cramp in non-XDP cases have been reported.27,28 The local anesthetic effect of lidocaine is expectedly fast but short acting, thus, the effects on the muscle may be prolonged by combining concentrated ethanol into the regimen (i.e., volume ratio of 10 : 1). MAB is a relatively safe procedure, however, patients complain of some dizziness and injection pain, secondary to ethanol effects. Ideally, it is better to monitor potential cardiac and central nervous effects of lidocaine, as quite a volume and dose of lidocaine are injected per muscle, with a number of muscles being targeted in a session.
- In 1995,29 we reported BoNT-A (onabotulinumtoxinA, Botox ®) effects in 5 cases of XDP having CD. Four weeks following a total of 200 units Botox®, the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS)30 showed marked improvement of the CD. This was followed by a larger series involving 25 cases of XDP with various types of CD.31 Following EMG-guided BoNT-A (abobotulinumtoxinA, Dysport®, dose range of 500–1,000 units) injections, we observed the following findings: 1) significant improvement in the TWSTRS scores 4 weeks post-injection; 2) significant VAS pain reduction 4 weeks post-injection, and 3) transient dysphagia in 4/25 cases, mainly in mixed CD given 1,000 units total dose of Dysport ®.31 BoNT-A blocks cholinergically innervated extrafusal and muscle spindle intrafusal fibers, thus modulating the sensory-motor circuitry of dystonia.32–34 Coupled to this phenomenon is the secondary effect of muscle weakening on the corresponding tendon and joint receptors.34
- From 1993 to 2003, we did a prospective, single-blind (independent scale assessors) study on 44 XDP cases, belonging to 24 families, to compare efficacy and safety of single-cycle BoNT-A versus MAB injections in CD.35 Consecutive XDP cases, irrespective of type of CD, were randomly distributed to the three interventions (i.e., Dysport®, Botox® and MAB). Per muscle EMG-guided injections of BoNT-A (Dysport® or D and Botox® or B) were as follows, respectively: For sternocleidomastoid, D=125 u (75–150 u); B=50 u (25–75 u); for splenius capitis, D=150 u (50–200 u); B= 60 u (40–80 u); for trapezius, D=100u (50–100 u); B= 40 u (30–60 u), and for levator scapula, D=50 u (25–75 u); B=20 u (10–30 u). A revised MAB protocol was used with lidocaine 1%, mixed with 95% ethanol (10 : 1 volume ratio), care being taken not to exceed 3–5 mg/kg body weight of total lidocaine injection per session. The primary outcome results comparing the 3 subscales of TWSTRS pre- and post-injection at week 4 indicated that both Dysport® and Botox® significantly improved in dystonia severity, disability and pain. MAB in CD did not show significant effects in any of the TWSTRS subscales. However, MAB showed modest improvement in the overall subjective dystonia rating scale at week 1, but this effect did not last longer than 2 weeks. Mean duration of the BoNT-A effects were in the order of 11 weeks, but not different between Dysport® and Botox®. Overall TWSTRS response scale of BoNT-A was significantly better than MAB, but not different between Dysport® and Botox®. Adverse events with BoNT-A were noted in weeks 1–3, and mainly in occasions of bilateral sternocleidomastoid injections for anterocollis. Dysphagia prevailed among the adverse events for BoNT-A (Dysport®: 3/15 cases; Botox®: 2/15; not significantly different from each other). For MAB, 8/14 XDP cases experienced nausea and dizziness, including pain on injection in 11/14 cases. Nevertheless, MAB is a cheap and rapid procedure, which can be performed while patients await funds for BoNT-A therapy.
- Injections with BoNT-A for disabling oromandibular, lingual and laryngeal dystonias should be performed cautiously, in view of the high rates of silent and overt aspiration in these XDP cases who may be in a malnourished state. The injection technique must require EMG guidance, expert choice of muscle targeting and BoNT-A low dosing with low dilution so as to prevent toxin spread. On the other hand, BoNT-A injections into the disabling limb and truncal dystonias of XDP may require higher toxin dosing and dilution to increase radius of spread and even reach those muscles with “overflow” of movements. From the aforementioned studies we performed, there seems to be a dichotomy of BoNT-A effects between the way the toxin relieves pain early and significantly compared to its effects upon the severe muscle spasms and dystonic posturing. It is also possible that, unlike cases of certain spasticity,36–38 early intervention with BoNT-A in dystonias of a neurodegenerative nature (e.g., XDP), may not be as beneficial.
- Surgery
- Data derived from the Philippine XDP registry (1960 to 1982), stereotactic ablative surgeries employing either staged bilateral pallidotomies or thalamotomies have been performed in 6 cases. Beset with the high rate of surgical complications, subsequent generations of Philippine XDP cases have been discouraged from seeking such surgical neuroablative procedures.39 Outside of the registry, some XDP cases underwent neuroablative procedures as well. For instance, a 34 year old Filipino who went through a right-sided cryothalamotomy for severe dystonia died 2 days postoperatively.40 Another patient treated with bilateral thalamotomy followed later by an implantation of a cerebellar stimulation device died of a postoperative cerebellar abscess.5
- Deep Brain Stimulation (DBS) functional surgery has the major advantage over neuroablative procedures for its reversibility and the avoidance of the common complications such as persistent dysarthria and cognitive impairment in bilateral lesioning procedures.41 Globus pallidus became the preferred empirical target for DBS in dystonia because of its proven effectiveness in ameliorating the off-dystonias and dyskinesias in Parkinson’s disease.42 To date, there have been 4 reports of GPi DBS done in XDP, showing favorable responses in all.43–46 To be added into the list are one case each of XDP who underwent GPi DBS performed in the Philippines39 and in Japan (unpublished). We had the opportunity to evaluate and follow up the latter two XDP cases, whose clinical presentations were those of focal dystonia that spread to other regions. The half-Japanese case had a Japanese father and a Filipina mother, whose ancestry is traced in Panay island. It generally appears that GPi DBS has promising results in ameliorating the disabling dystonia of these patients and minus the persistent adverse events.
Challenges in therapy
- Herein, we depict XDP in a family, highlighting the challenging aspects of the disease from phenomenology, therapy, social impact and his eventual demise. The illustrative case is a 44 year old male XDP, whose maternal ancestry belonged to Roxas city of Capiz, and whose manifestations and course were shortened. His elder brother in a brood of 5 (4 males, one female in a family proband) was also diagnosed to have XDP. This elder brother had an onset of oromandibular and cervical dystonia at age 40 years. The dystonia eventually became generalized at age 44 years of this brother. On our examination at age 45 years, this brother also had rest tremors and bradykinesia, in addition to the oromandibulo-lingual, cervical, truncal and limb dystonias. Unable to continue employment as a seafarer and “stigmatized” at his hometown, he isolated himself. At age 47 years, this brother committed suicide one day, by hanging himself, having lost hope due to severe, disabling symptoms with relentless progression. A maternal uncle was also diagnosed to have XDP (though not examined by us, we had information that he had cervical and jaw-opening and tongue protusion dystonias) at age 40 years. This maternal uncle succumbed to aspiration pneumonia at age 51 years, barely able to feed and had become slowed in movements.
- In regard to the illustrative case, he initially developed focal upper limb dystonia, with right wrist extension at age 39 years. Being right-handed and an able-bodied seaman, the focal dystonia started to impede work. The first consult was triggered by the focal upper limb dystonia and localized pain in the same area, physical trauma having been ruled out. His wrist extensors initially responded to MAB with lidocaine-ethanol but efficacy would only last 2 weeks. Thus, BoNT-A was injected to the extensor carpi ulanris/radialis (each with 100 Dysport®) and extensor digitorum communis (75 units Dysport ®), providing significant relief of muscle spasms, posturing and especially pain for at least 3 months. A year later, he developed blepharospasm and jaw-closing dystonia, which together with the limb dystonia, again responded well to BoNT-A. In the absence of available tetrabenezine and trihexyphenidyl, dose escalating doses of clonazepam was started (initially at 0.5 mg/day and moved up to 4 mg/day) and later combined with an anticholinergic agent (biperiden, final total dose of 12 mg/day). Baclofen (up to 30 mg/day) was added into the regimen on his 3rd year, when dystonia became generalized. However, tolerability issues led him to retain the clonazepam and biperiden doses only. These oral medications became the maintenance medications over time, as he could not fend for the increasing doses BoNT-A injections. He was unable to maintain his seafaring job that led to his depression. Sertraline (up to 20 mg/day) was tried for 3 months but was eventually discontinued, as he noted confounding sedative and intolerance effects of the medication. On his 3rd year of the illness, he noted slowness in movements and periodic slow rhythmic rest hand tremors, in addition to the generalized dystonia. Levodopa-carbidopa (final total dose of 750 mg/day) provided initial relief of the parkinsonism features, but this same medication was discontinued because of eventual loss of efficacy. The course of the illness changed, when he was admitted as an emergency for “status dystonicus,” with severe generalized dystonia, including involvement of his respiratory muscles and laryngeal stridor. His serial total creatine kinase levels were elevated 10× the normal, an elevated creatinine (3× the normal) and hemoglobinuria. Diazepam bolus followed by an intravenous drip (total 60 mg/day) was given and was gradually reduced as his dystonic spasms lessened. Tracheostomy was performed, whilst requiring a respirator. The wife reported that the patient lost hope of the possibility of DBS for financial reasons, that lead to his abrupt discontinuation of his cloanzepam and biperiden, five days prior to his confinement. In the hospital, zolpidem monotherapy was initiated and escalated (initially at 10 mg/day and moved up to 30 mg/day in 3 divided doses), that appeared to have improved his dystonia. He was gradually weaned from the respirator and maintained on zolpidem upon discharge after two months in the hospital. He apparently tolerated over a period of 4 weeks the daytime sleepiness associated with zolpidem. Two weeks from discharge, we learned that the patient succumbed in his sleep as he purposely refused to continue treatment, having lost any hope of cure and “catastrophized” by the moribund nature of the illness.
Illustrative case
- Whilst the clinical phenotype of XDP appears to be straightforward, certain gaps need to be resolved in the future. Phenomenologically, XDP cases in families have been identified whose ancestries cannot be traced from the Panay island. Variability of disease onset, clinical manifestations and disease duration between affected members within families require a revisit into the XDP phenotype. Occurrence of manifesting females with XDP will require a revisit to the genetic profile of the disease. /DYT3/ is still a putative gene at the moment and the protein product has not been purified. At the moment, in depth studies into the function of /TAF1/ and how it leads to disease need to be elaborated. Apart from expense limiting DBS, new pharmacotherapeutic strategies are desired in view of the shortcomings of current regimen for dystonia, especially when XDP enters into a combined phase with parkinsonism. Finally, there is urgency for us to address these issues because our XDP cases are losing hope and have started to take their lives away themselves.
Conclusion
- Dr. Lillian Lee provided valuable advices and served as a great inspiration as the author embarked on projects dedicated to XDP. Drs. D. Jamora, A. Ng, P. Pasco, C. Diesta, S. Teleg, E. Munoz, J. Aguilar and S. Sarcia lent their expertise, while MJ Monding assisted in several XDP projects.
Acknowledgments
Acknowledgments
-
Declaration of Interest
Dr. Rosales does consulting agreements and speaking engagements for Ipsen Pharmaceuticals. He has performed clinical researches sponsored by Ipsen and Allergan through a contract with the University of Santo Tomas and the CNS of Metropolitan Medical Center in Manila. As president of the Movement Disorders Society of the Philippines, he has worked with and developed collaborative projects with the XDP study group, which are intended for the XDP cause. The author alone is responsible for the content and writing of the manuscript.
Notes
- 1. Lee LV, Pascasio FM, Fuentes FD, Viterbo GH. Torsion dystonia in Panay, Philippines. Adv Neurol 1976;14:137–151.PubMed
- 2. Lee LV, Rivera C, Teleg RA, Dantes MB, Pasco PM, Jamora RD, et al. The Unique Phenomenology of Sex-Linked Dystonia Parkinsonism (XDP, DYT3, “Lubag”). Int J Neurosci In press. Article
- 3. Lee LV, Kupke KG, Caballar-Gonzaga F, Hebron-Ortiz M, Müller U. The phenotype of the X-linked dystonia-parkinsonism syndrome. An assessment of 42 cases in the Philippines. Medicine (Baltimore) 1991;70:179–187.ArticlePubMed
- 4. Lee LV, Munoz EL, Tan KT, Reyes MT. Sex linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Mol Pathol 2001;54:362–368.ArticlePubMedPMC
- 5. Lee LV, Maranon E, Demaisip C, Peralta O, Borres-Icasiano R, Arancillo J, et al. The natural history of sex-linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Parkinsonism Relat Disord 2002;9:29–38.ArticlePubMed
- 6. Evidente VG, Nolte D, Niemann S, Advincula J, Mayo MC, Natividad FF, et al. Phenotypic and molecular analyses of X-linked dystonia-parkinsonism (‘lubag’) in women. Arch Neurol 2004;61:1956–1959.ArticlePubMed
- 7. Lew MF, Shindo M, Moskowitz CB, Wilhelmsen KC, Fahn S, Waters CH. Adductor laryngeal breathing dystonia in a patient with lubag (X-linked dystonia-Parkinsonism syndrome). Mov Disord 1994;9:318–320.ArticlePubMed
- 8. Evidente VG, Advincula J, Esteban R, Pasco P, Alfon JA, Natividad FF, et al. Phenomenology of “Lubag” or X-linked dystonia-parkinsonism. Mov Disord 2002;17:1271–1277.ArticlePubMed
- 9. Evidente VG, Gwinn-Hardy K, Hardy J, Hernandez D, Singleton A. X-linked dystonia (“Lubag”) presenting predominantly with parkinsonism: a more benign phenotype? Mov Disord 2002;17:200–202.ArticlePubMed
- 10. Evidente VG. 2010 http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=xdp.
- 11. Pasco PM, Ison CV, Magpusao NS, Muňoz EL, Reyes MT. Understanding XDP through imaging, pathology, and genetics. Int J Neurosci In press. Article
- 12. Evidente VG, Dickson D, Singleton A, Natividad F, Hardy J, Hernandez D. Novel neuropathological findings in Lubag or x-linked dystonia-parkinsonism. Mov Disord 2002;17( Suppl 1):294.
- 13. Goto S, Lee LV, Munoz EL, Tooyama I, Tamiya G, Makino S, et al. Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann Neurol 2005;58:7–17.ArticlePubMed
- 14. Nolte D, Niemann S, Müller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A 2003;100:10347–10352.ArticlePubMedPMC
- 15. Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet 2007;80:393–406.ArticlePubMedPMC
- 16. Deng H, Le WD, Jankovic J. Genetic study of an American family with DYT3 dystonia (lubag). Neurosci Lett 2008;448:180–183.ArticlePubMed
- 17. Kupke KG, Lee LV, Müller U. Assignment of the X-linked torsion dystonia gene to Xq21 by linkage analysis. Neurology 1990;40:1438–1442.ArticlePubMed
- 18. Waters CH, Takahashi H, Wilhelmsen KC, Shubin R, Snow BJ, Nygaard TG, et al. Phenotypic expression of X-linked dystonia-parkinsonism (lubag) in two women. Neurology 1993;43:1555–1558.ArticlePubMed
- 19. Gordon MF, Diaz-Olivo R, Hunt AL, Fahn S. Therapeutic trial of milacemide in patients with myoclonus and other intractable movement disorders. Mov Disord 1993;8:484–488.ArticlePubMed
- 20. Langer SZ, Arbilla S, Scatton BV, Niddam R, Dubois A. Receptors involved in the mechanisms of action of zolpidem. In: Sauvanet JP, Langer SZ, Morselli PL, editors. Imidazopyridines in Sleep Disorders. New York: Raven Press; 1988:5–70.
- 21. Chen YY, Sy HN, Wu SL. Zolpidem improves akinesia, dystonia and dyskinesia in advanced Parkinson’s disease. J Clin Neurosci 2008;15:955–956.ArticlePubMed
- 22. Daniele A, Moro E, Bentivoglio AR. Zolpidem in progressive supranuclear palsy. N Engl J Med 1999;341:543–544.Article
- 23. Rüzicka E, Roth J, Jech R, Busek P. Subhypnotic doses of zolpidem oppose dopaminergic-induced dyskinesia in Parkinson’s disease. Mov Disord 2000;15:734–735.ArticlePubMed
- 24. Evidente VG. Zolpidem improves dystonia in “Lubag” or X-linked dystonia-parkinsonism syndrome. Neurology 2002;58:662–663.ArticlePubMed
- 25. Auta J, Impagnatiello F, Kadriu B, Guidotti A, Costa E. Imidazenil: a low efficacy agonist at α1-but high efficacy at α5-GABA-A receptors fail to show anticonvulsant cross tolerance to diazepam or zolpidem. Neuropharmacol 2008;55:148–153.ArticlePubMedPMC
- 26. Teleg R, Dantes M, Peralta O, Icasiano R, Lee LV. Muscle Afferent Block in the Treatment of X-linked Dystonia-Parkinsonism (XDP). Acta Med Phil 2005;39:28–35.
- 27. Kaji R, Rothwell JC, Katayama M, Ikeda T, Kubori T, Kohara N, et al. Tonic vibration reflex and muscle afferent block in writer’s cramp. Ann Neurol 1995;38:155–162.ArticlePubMed
- 28. Kaji R. Muscle afferent block for dystonia: Implications for the physiological mechanism of action of botulinum toxin. Scientific and Therapeutic Aspects of Botulinum Toxin. In: Brin MF, Jankovic J, Hallett M, editors. Philadelphia: Lippincott, Williams & Wilkins; 2002:455–458.
- 29. Rosales RL, Arimura K, Gamez G, Osame M. X-linked dystonia-parkinsonism: botulinum toxin therapy and stimulation single-fiber electromyography. Electroencephal Clin Neurophysiol (Section of Electromyography and Motor Control)(Suppl) 1995;97:S234–235.Article
- 30. Consky ES, Basinski A, Belle L, Ranawaya R, Lang AE. The Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS): assessment of validity and inter-rater reliability. Neurology 1990;40:445.
- 31. Rosales RL, Delgado MS, Rosales ME. Spasticity, X-linked Dystonia, Parkinsonism and Botulinum Toxin A: The Local Scenario. Proceedings of the 3rd World Congress of Neurological Rehabilitation. In: Battistin L, Dam M, Tonin P, editors. Italy: Monduzzi Editore; 2002:121–1260.
- 32. Rosales RL, Arimura K, Takenaga S, Osame M. Extrafusal and intrafusal muscle effects in experimental botulinum toxin-A injection. Muscle Nerve 1996;19:488–496.ArticlePubMed
- 33. Rosales RL, Bigalke H, Dressler D. Pharmacology of botulinum toxin: differences between type A preparations. Eur J Neurol 2006;13( Suppl 1):2–10.Article
- 34. Rosales RL, Dressler D. On Muscle spindles, dystonia and botulinum toxin. Eur J Neurol 2010;17:71–80.ArticlePubMed
- 35. Rosales RL, Delgado M, Malicdan M. Botulinum Toxin A and Muscle Afferent block in X-linked Dystonia-Parkinsonism of Panay. Mov Disord (Suppl 9):2004;19S103–104.
- 36. Rosales RL, Chua-Yap AS. Evidence-based systematic review on the efficacy and safety of botulinum toxin-A therapy in post-stroke spasticity. J Neural Transm 2008;115:617–623.ArticlePubMed
- 37. Rosales RL, Kong HK. for the Asian Botulinum toxin study group. The Asian botulinum toxin-A (Dysport) clinical trial designed for early post-stroke spasticity (ABCDE-S trial). Mov Disord (Suppl) 2009;24:S455.ArticlePubMedPMC
- 38. Lukban MB, Rosales RL, Dressler D. Effectiveness of botulinum toxin A for upper and lower limb spasticity in children with cerebral palsy: a summary of evidence. J Neural Transm 2009;116:319–331.ArticlePubMed
- 39. Aguilar JA, Vesagas TS, Jamora RD, Teleg RA, Ledesma L, Rosales RL, et al. The Promise of Deep Brain Stimulation in X-Linked Dystonia Parkinsonism. Int J Neurosci In press. Article
- 40. Waters CH, Faust PL, Powers J, Vinters H, Moskowitz C, Nygaard T, et al. Neuropathology of lubag (x-linked dystonia parkinsonism). Mov Disord 1993;8:387–390.ArticlePubMed
- 41. Vidailhet M, Vercueil L, Houeto JL, Krystkowiak P, Benabid AL, Cornu P, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med 2005;352:459–467.ArticlePubMed
- 42. Greene P. Deep-brain stimulation for generalized dystonia. N Engl J Med 2005;352:498–500.ArticlePubMed
- 43. Evidente VG, Lyons MK, Wheeler M, Hillman R, Helepolelei L, Beynen F, et al. First case of X-linked dystonia-parkinsonism (“Lubag”) to demonstrate a response to bilateral pallidal stimulation. Mov Disord 2007;22:1790–1793.ArticlePubMed
- 44. Martinez-Torres I, Limousin P, Tisch S, Page R, Pinto A, Foltynie T, et al. Early and marked benefit with GPi DBS for Lubag syndrome presenting with rapidly progressive life-threatening dystonia. Mov Disord 2009;24:1710–1712.ArticlePubMed
- 45. Wadia PM, Lim SY, Lozano AM, Adams JR, Poon YY, Diaz CT, et al. Bilateral pallidal stimulation for x-linked dystonia parkinsonism. Arch Neurol 2010;67:1012–1015.ArticlePubMed
- 46. Oyama G, Fernandez HH, Foote KD, Zeilman P, Hwynn N, Jacobson CE 4th, et al. Differential response of dystonia and parkinsonism following globus pallidus internus deep brain stimulation in X-linked dystonia-parkinsonism (Lubag). Stereotact Funct Neurosurg 2010;88:329–333.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- A scoping review on the diagnosis and treatment of X-linked dystonia-parkinsonism
Anisah Hayaminnah D. Alonto, Roland Dominic G. Jamora
Parkinsonism & Related Disorders.2024; 119: 105949. CrossRef - Progressive Decline in Voice and Voice-Related Quality of Life in X-Linked Dystonia Parkinsonism
Sungjin A. Song, Criscely L. Go, Patrick B. Acuna, Jan Kristopher Palentinos De Guzman, Nutan Sharma, Phillip C. Song
Journal of Voice.2023; 37(1): 134. CrossRef - X-linked dystonia parkinsonism: epidemiology, genetics, clinical features, diagnosis, and treatment
Hok Leong Chin, Chia-Yi Lin, Oscar Hou-In Chou
Acta Neurologica Belgica.2023; 123(1): 45. CrossRef - Basal Ganglia Atrophy as a Marker for Prodromal X‐Linked Dystonia‐Parkinsonism
Henrike Hanssen, Cid C. E. Diesta, Marcus Heldmann, Jackson Dy, Jeffrey Tantianpact, Julia Steinhardt, Rosanna Sauza, Hans T. S. Manalo, Andreas Sprenger, Charles Jourdan Reyes, Raphael Tuazon, Björn‐Hergen Laabs, Aloysius Domingo, Raymond L. Rosales, Chr
Annals of Neurology.2023; 93(5): 999. CrossRef - Oculomotor abnormalities indicate early executive dysfunction in prodromal X-linked dystonia-parkinsonism (XDP)
Renana Mertin, Cid Diesta, Norbert Brüggemann, Raymond L. Rosales, Henrike Hanssen, Ana Westenberger, Julia Steinhardt, Marcus Heldmann, Hans T. S. Manalo, Jean Q. Oropilla, Christine Klein, Christoph Helmchen, Andreas Sprenger
Journal of Neurology.2023; 270(9): 4262. CrossRef - Endemic parkinsonism: clusters, biology and clinical features
Katerina Menšíková, John C. Steele, Raymond Rosales, Carlo Colosimo, Peter Spencer, Annie Lannuzel, Yoshikazu Ugawa, Ryogen Sasaki, Santiago Giménez-Roldán, Radoslav Matej, Lucie Tuckova, Dominik Hrabos, Kristyna Kolarikova, Radek Vodicka, Radek Vrtel, Mi
Nature Reviews Neurology.2023; 19(10): 599. CrossRef - Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing
Theresa Lüth, Joshua Laβ, Susen Schaake, Inken Wohlers, Jelena Pozojevic, Roland Dominic G. Jamora, Raymond L. Rosales, Norbert Brüggemann, Gerard Saranza, Cid Czarina E. Diesta, Kathleen Schlüter, Ronnie Tse, Charles Jourdan Reyes, Max Brand, Hauke Busch
Genes.2022; 13(1): 126. CrossRef - Prodromal X‐Linked Dystonia‐Parkinsonism is Characterized by a Subclinical Motor Phenotype
Julia Steinhardt, Henrike Hanssen, Marcus Heldmann, Andreas Sprenger, Björn‐Hergen Laabs, Aloysius Domingo, Charles Jourdan Reyes, Jannik Prasuhn, Max Brand, Raymond Rosales, Thomas F. Münte, Christine Klein, Ana Westenberger, Jean Q. Oropilla, Cid Diesta
Movement Disorders.2022; 37(7): 1474. CrossRef - A Community-based study on the prevalence and predisposing factors of Parkinson’s disease in Barangay Mangilag Sur, Quezon Province, Philippines
Raymond L. Rosales, Mary Camille E. Rosales, Danica Jane S.J. Robles, Ron Christian Neil T. Rodriguez, Nadia Beatrice S. Romana, Joseph Mariuz B. Rosales, Gerardo B. Salazar, Richelle Ann S. Santiano
Clinical Parkinsonism & Related Disorders.2022; 7: 100169. CrossRef - Retroelement-derived RNA and its role in the brain
Taylor A. Evans, Jennifer Ann Erwin
Seminars in Cell & Developmental Biology.2021; 114: 68. CrossRef - Behandlungsstrategien bei oromandibulärer Dystonie
Kazuya Yoshida
Fortschritte der Neurologie · Psychiatrie.2021; 89(11): 562. CrossRef - X-linked dystonia Parkinsonism: crossing a new threshold
Arlene R. Ng, Roland Dominic G. Jamora, Raymond L. Rosales
Journal of Neural Transmission.2021; 128(4): 567. CrossRef - A Cross-Cultural Validation of the Filipino and Hiligaynon Versions of the Parts IIIB (Non-Motor Features) and IV (Activities of Daily Living) of the X-Linked Dystonia-Parkinsonism– MDSP Rating Scale
Richelle Ann S. Santiano, Raymond L. Rosales
Clinical Parkinsonism & Related Disorders.2021; 5: 100100. CrossRef - Speech and swallowing deficits in X-Linked Dystonia-Parkinsonism
Ana Luiza Zaninotto, Jan K. de Guzman, Kaila L. Stipancic, Bridget J. Perry, Melanie L. Supnet, Criscely Go, Nutan Sharma, Jordan R. Green
Parkinsonism & Related Disorders.2021; 89: 105. CrossRef - Tremor in Primary Monogenic Dystonia
Sanjay Pandey, Sonali Bhattad, Shreya Dinesh
Current Neurology and Neuroscience Reports.2021;[Epub] CrossRef - Oromandibular Dystonia: A Clinical Examination of 2,020 Cases
Laura M. Scorr, Stewart A. Factor, Sahyli Perez Parra, Rachel Kaye, Randal C. Paniello, Scott A. Norris, Joel S. Perlmutter, Tobias Bäumer, Tatiana Usnich, Brian D. Berman, Marie Mailly, Emmanuel Roze, Marie Vidailhet, Joseph Jankovic, Mark S. LeDoux, Ric
Frontiers in Neurology.2021;[Epub] CrossRef - Voice and swallowing dysfunction in X‐linked dystonia parkinsonism
Phillip C. Song, Hoai Le, Patrick Acuna, Jan Kristopher Palentinos De Guzman, Nutan Sharma, Taylor N. Francouer, Marisela E. Dy, Criscely L. Go
The Laryngoscope.2020; 130(1): 171. CrossRef - Increased insula-putamen connectivity in X-linked dystonia-parkinsonism
Anne J. Blood, Jeff L. Waugh, Thomas F. Münte, Marcus Heldmann, Aloysius Domingo, Christine Klein, Hans C. Breiter, Lillian V. Lee, Raymond L. Rosales, Norbert Brüggemann
NeuroImage: Clinical.2018; 17: 835. CrossRef - Altered glutamate response and calcium dynamics in iPSC-derived striatal neurons from XDP patients
P. Capetian, N. Stanslowsky, E. Bernhardi, K. Grütz, A. Domingo, N. Brüggemann, M. Naujock, P. Seibler, C. Klein, F. Wegner
Experimental Neurology.2018; 308: 47. CrossRef - Eye movement deficits in X-linked dystonia-parkinsonism are related to striatal degeneration
Andreas Sprenger, Henrike Hanssen, Imke Hagedorn, Jannik Prasuhn, Raymond L. Rosales, Roland Dominic G. Jamora, Cid C. Diesta, Aloysius Domingo, Christine Klein, Norbert Brüggemann, Christoph Helmchen
Parkinsonism & Related Disorders.2018;[Epub] CrossRef - Validation of a screening questionnaire for X‐linked dystonia parkinsonism: The first phase of the population‐based prevalence study of X‐linked dystonia parkinsonism in Panay
Jose Danilo B Diestro, Paul Matthew D Pasco, Lillian V Lee
Neurology and Clinical Neuroscience.2017; 5(3): 79. CrossRef - Validation of the XDP–MDSP rating scale for the evaluation of patients with X-linked dystonia-parkinsonism
Paul Matthew D. Pasco, Roland Dominic G. Jamora, Raymond L. Rosales, Cid Czarina E. Diesta, Arlene R. Ng, Rosalia A. Teleg, Criscely L. Go, Lillian Lee, Hubert H. Fernandez
npj Parkinson's Disease.2017;[Epub] CrossRef - Clinicopathological Phenotype and Genetics of X-Linked Dystonia–Parkinsonism (XDP; DYT3; Lubag)
Toshitaka Kawarai, Ryoma Morigaki, Ryuji Kaji, Satoshi Goto
Brain Sciences.2017; 7(12): 72. CrossRef - Prevalence and Predisposing Factors of Parkinson Disease: A Community-Based Study In Barangay Mangilag Sur, Candelaria, Quezon: A Research Protocol
Danica Jane S.J Robles, Ron Christian Neil T Rodriguez, Nadia Beatrice S Romana, Joseph Mariuz B Rosales, Mary Camille E Rosales, Gerardo B Salazar, Raymond L Rosales
Journal of Medicine, University of Santo Tomas.2017; 1(1): 109. CrossRef - Evidence of TAF1 dysfunction in peripheral models of X-linked dystonia-parkinsonism
Aloysius Domingo, David Amar, Karen Grütz, Lillian V. Lee, Raymond Rosales, Norbert Brüggemann, Roland Dominic Jamora, Eva Cutiongco-dela Paz, Arndt Rolfs, Dirk Dressler, Uwe Walter, Dimitri Krainc, Katja Lohmann, Ron Shamir, Christine Klein, Ana Westenbe
Cellular and Molecular Life Sciences.2016; 73(16): 3205. CrossRef - Can a Positive Allosteric Modulation of GABAergic Receptors Improve Motor Symptoms in Patients with Parkinson’s Disease? The Potential Role of Zolpidem in the Treatment of Parkinson’s Disease
Antonio Daniele, Francesco Panza, Antonio Greco, Giancarlo Logroscino, Davide Seripa
Parkinson's Disease.2016; 2016: 1. CrossRef - Introduction
Erle C.H. Lim, Roongroj Bhidayasiri, Raymond L. Rosales, Ryuji Kaji
Parkinsonism & Related Disorders.2011; 17: S1. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite