E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(3); 2018 > Article
-
Case Report
A Patient with Myotonic Dystrophy Type 1 Presenting as Parkinsonism -
Ji-Hyun Choi1,2, Jee-Young Lee2, Han-Joon Kim1, Beomseok Jeon1

-
Journal of Movement Disorders 2018;11(3):145-148.
DOI: https://doi.org/10.14802/jmd.18028
Published online: September 30, 2018
1Department of Neurology and Movement Disorder Center, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
2Department of Neurology, Seoul Metropolitan Government–Seoul National University Boramae Medical Center, Seoul National University College of Medicine, Seoul, Korea
- Corresponding author: Beomseok Jeon, MD, PhD, https://orcid.org/0000-0003-2491-3544 Department of Neurology, Seoul National University Hospital, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea / Tel: +82-2-2072-2876 / Fax: +82-2- 3672-7553 / E-mail: brain@snu.ac.kr
• Received: June 8, 2018 • Revised: July 3, 2018 • Accepted: July 24, 2018
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 5,753 Views
- 103 Download
- 1 Web of Science
ABSTRACT
- The current body of literature contains 5 reports of myotonic dystrophy (DM) with parkinsonism: 4 reports of DM type 2 and 1 report of clinically suspected DM type 1. To date, there have been no genetically proven cases of DM type 1 with parkinsonism. Here, we report the first case of genetically proven DM type 1 and parkinsonism that developed ahead of muscle symptoms with bilateral putaminal, presynaptic dopaminergic deficits on imaging. A 54-year-old female patient presented with bradykinesia, axial and bilateral limb rigidity, stooped posture, and hypomimia, which did not respond to levodopa. At age 56, she developed neck flexion weakness. Examination showed bilateral facial weakness, percussion and grip myotonia, and electromyography confirmed myotonic discharges. A genetic study of DM type 1 showed a DMPK mutation. At age 58, gait freezing, postural instability, and frequent falling developed and did not respond to increasing doses of levodopa. At age 59, the patient died from asphyxia.
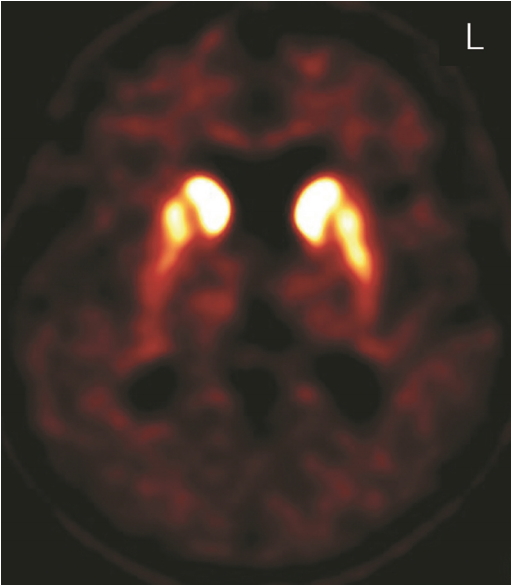
- A 54-year-old female presented with complaints of a oneyear history of uncomfortable range of motion in all four limbs with stiffness and slowness while moving and walking, and she had no significant history of prior illness or medication. Family history revealed that one of her three siblings experienced a cerebral infarction at the age of 52, and her mother was diagnosed with dementia at the age of 90. On initial examination at age 54, the patient demonstrated hypomimia, mild to moderate bilateral bradykinesia on finger tapping, hand-grasp, and pronation-supination movements of the hands, mild axial and bilateral limb rigidity, mildly stooped posture, and slowing gait with bilaterally reduced arm swing, though no resting or kinetic tremor. Eye movements showed breaking up of smooth pursuit but no supranuclear gaze palsy. Autonomic symptoms and signs were not evident. She did not have depression, sleep problems, or loss of smell. Her Mini-Mental State Examination score was 29/30. The Frontal Assessment Battery score was 15/18, which suggested mild frontal lobe dysfunction. MRI of the brain showed mild diffuse cerebral atrophy. Serum copper and ceruloplasmin tests for Wilson disease were within the normal ranges. A levodopa trial showed subjective minimal improvement. The initial diagnosis was atypical parkinsonian syndrome based on bilateral symmetric parkinsonian features and poor levodopa response. DAT imaging using an (18)F-FP-CIT [(18)F-fluorinated N-3-fluoropropyl-2-beta-carboxymethoxy-3-beta-(4-iodophenyl) nortropane] positron emission tomography scan showed moderately to severely decreased uptake in the putamen bilaterally (Figure 1). At age 56, she developed difficulties raising her head up when lying on her back. Examination showed neck flexion weakness, mild bilateral facial weakness, and percussion and grip myotonia. Needle electromyography confirmed myotonic discharges in proximal and distal muscles. Ophthalmologic examination showed bilateral cataracts. Echocardiography and electrocardiography were normal. A genetic screen for DM type 1 showed an abnormal CTG repeat length with 120 repeats in the untranslated 3' region of DMPK on chromosome 19q13.3. By age 58, the patient had developed gait freezing, postural instability, and frequent falling, as well as progressed to moderate to severe bilateral bradykinesia on finger tapping, hand-grasp, and pronation-supination movements of the hands, moderate to severe axial and bilateral limb rigidity, and moderate stooped posture. Increasing her dose of levodopa to 1,300 mg per day was not effective. She developed dysarthria, dysphasia, and severe weight loss. At age 59, she died from asphyxia. An autopsy was not performed.
CASE REPORT
- In this report, the patient was initially diagnosed with atypical parkinsonian syndrome based on bilateral symmetric parkinsonian features and a poor levodopa response. DAT imaging confirmed the presynaptic dopaminergic deficits in the bilateral putamen. Later, she was diagnosed with DM type 1 based on myotonia clinical manifestations and DMPK mutations. To date, there have been no reports of genetically proven cases of DM type 1 with parkinsonism. To our knowledge, this is the first reported case of DM type 1 with DMPK mutations and parkinsonism that developed ahead of muscle symptoms with bilateral putaminal, presynaptic dopaminergic deficits on DAT imaging. The prevalence of DM type 1 appears to be more common than DM type 2, except in Germany, Poland, and Finland, where DM type 2 is as common as DM type 1 [1]. However, parkinsonism has been reported more often in DM type 2 cases than in type 1 cases (Table 1).
- Of the previous 5 reported patients with DM who developed parkinsonism (Table 1), an autopsy was performed on 1 patient with clinically diagnosed DM type 1 and parkinsonism [2], who was never genetically tested. The autopsy in the patient showed marked cell loss and Lewy bodies in the substantia nigra (SN) and diffuse myelin loss in the cerebral white matter [2]. However, Lewy bodies in the SN have been reported in other patients with genetically proven or clinically diagnosed DM without parkinsonism [7]. Other neuropathological changes found upon autopsy of genetically proven or clinically diagnosed DM included neurofibrillary tangles, observed mostly in the hippocampus and the temporal neocortex [8]. Marinesco bodies and neuronal loss of the medullary arcuate nucleus and reticular formation were also noted in some cases [9], but were not found consistently, nor were clinicopathological features consistent. Other neuropathological findings included intracytoplasmic inclusion bodies in the thalamus, SN, cerebral cortex, caudate nucleus, and putamen [10], of which the mechanism of pathology remains unknown. Lewy bodies and intracytoplasmic inclusion bodies found in the SN or the subcortical striatum may raise the possibility of association between parkinsonism and DM, but it warrants further studies.
- In conclusion, parkinsonism has been reported in patients with DM. The association of parkinsonism and DM may not be coincidental given the current case and other similar reports and their neuropathology results. However, further research is needed to understand the relationship between parkinsonism symptoms and DM.
DISCUSSION
- The DNA sample for this study was provided by the Seoul National University Hospital Human Biobank, a member of the National Biobank of Korea, which is supported by the Ministry of Health and Welfare. All samples derived from the National Biobank of Korea were obtained with informed consent under institutional review board-approved protocols.
Acknowledgments
Figure 1.FP-CIT positron emission tomography scan showed moderately to severely decreased uptake in the putamen bilaterally.
Table 1.Summary of patients with DM and parkinsonism
| Authors |
Patients |
DM features |
Parkinsonian features |
|||||
|---|---|---|---|---|---|---|---|---|
| Age (yrs) | Sex | Gene test for DM | Diagnosed DM type | Onset age of muscle involvement (yrs) | Onset age of parkinsonism (yrs) | Parkinsonism | Levodopa response for parkinsonism | |
| Okuma et al. [2] | 63 | F | Not performed | Type 1 | 40 | 50 | Asymmetric tremor, small step with freezing, bradykinesia, postural instability and hypomimia | Good response |
| Hund et al. [3] | 59 | F | DM type 2 gene positive | Type 2 | 50 | 52 | Stooped posture, hypomimia and low voice | Good response |
| Chu et al. [4] | 65 | M | DM type 1 gene negative not performed for type 2 | Type 2 | 55 | 63 | Asymmetrical tremor, rigidity, bradykinesia, hypomimia, stooped posture, and gait disturbance | Partial response |
| Celik et al. [5] | 41 | F | Not performed | Type 2 | 38 | 40 | Bilateral resting tremor, rigidity, bradykinesia and hypomimia | Good response |
| Annic et al. [6] | 47 | F | DM type 2 gene positive | Type 2 | 51 | 47 | Bilateral tremor, bradykinesia, rigidity and gait disturbance | Partial response |
| Current case | 54 | F | DM type 1 gene positive | Type 1 | 56 | 53 | Bilateral bradykinesia, rigidity, stooped posture, hypomimia, gait freezing and postural instability | Minimal response |
- 1. Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 2012;11:891–905.ArticlePubMed
- 2. Okuma Y, Tanaka S, Nomura Y, Mori H, Yan H, Shirai T, et al. [A 63-year-old woman with muscle weakness, myotonia, and parkinsonism]. No To Shinkei 1996;48:287–297.PubMed
- 3. Hund E, Jansen O, Koch MC, Ricker K, Fogel W, Niedermaier N, et al. Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology 1997;48:33–37.ArticlePubMed
- 4. Chu K, Cho JW, Song EC, Jeon BS. A patient with proximal myotonic myopathy and parkinsonism. Can J Neurol Sci 2002;29:188–190.ArticlePubMed
- 5. Celik Y, Turgut N, Balci K, Kabayel L. Proximal myotonic dystrophy associated with parkinsonism. J Clin Neurosci 2006;13:275–276.ArticlePubMed
- 6. Annic A, Devos D, Destée A, Defebvre L, Lacour A, Hurtevent JF, et al. Early dopasensitive Parkinsonism related to myotonic dystrophy type 2. Mov Disord 2008;23:2100–2101.ArticlePubMed
- 7. Itoh K, Mitani M, Kawamoto K, Futamura N, Funakawa I, Jinnai K, et al. Neuropathology does not correlate with regional differences in the extent of expansion of CTG repeats in the brain with myotonic dystrophy type 1. Acta Histochem Cytochem 2010;43:149–156.ArticlePubMedPMC
- 8. Maurage CA, Udd B, Ruchoux MM, Vermersch P, Kalimo H, Krahe R, et al. Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology 2005;65:1636–1638.ArticlePubMed
- 9. Ono S, Takahashi K, Kanda F, Jinnai K, Fukuoka Y, Mitake S, et al. Decrease of neurons in the medullary arcuate nucleus in myotonic dystrophy. Acta Neuropathol 2001;102:89–93.ArticlePubMedPDF
- 10. Ono S, Takahashi K, Fukuoka Y, Jinnai K, Kanda F, Kurisaki H, et al. Intracytoplasmic inclusion bodies of the substantia nigra in myotonic dystrophy. Immunohistochemical observations. J Neurol Sci 1997;148:193–198.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite