1Division of Neurology, Department of Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
2The Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, University of Malaya, Kuala Lumpur, Malaysia
Corresponding author: Ai Huey Tan, MD, MRCP (UK), https://orcid.org/0000-0002-2979-3839 Neurology Laboratory, Level 6 (South Block), University of Malaya Medical Centre, 50603 Kuala Lumpur, Malaysia / Tel: +60-3-7949-2891 / Fax: +60- 3-7949-4613 / E-mail: aihuey.tan@gmail.com
• Received: March 24, 2018 • Revised: April 16, 2018 • Accepted: April 24, 2018
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Creutzfeld-Jakob disease (CJD) is a rare neurodegenerative disease that is clinically characterized by rapidly progressive dementia, visual disturbance, and movement disorders including myoclonus and cerebellar ataxia, both of which occur in approximately 80% of patients [1]. Over time, almost all patients enter a phase of akinetic mutism before death. Although chorea is commonly observed in variant CJD (vCJD), it is considered a rare occurrence in sporadic CJD (sCJD) [2]. Here, we report a patient with sCJD with chorea as a prominent clinical feature on presentation to the hospital.
A previously healthy 71-year-old woman presented with a two-month history of visual hallucinations followed by rapidly worsening cognitive function and mobility. Within six weeks of symptom onset, she became completely withdrawn and unable to recognize family members or respond to questions; her gait also progressed from imbalance requiring assistance to a bedridden state. At this point, she was noted to have abnormal movements. There was no history of fever or constitutional symptoms. The patient had travelled to Vietnam two months prior to symptom onset, but there was no other history of overseas travel. There was no relevant family history.
The patient was referred to our center two months after first symptom onset. On admission, she was unable to obey commands and produced only incomprehensible sounds. She had generalized chorea involving the face, trunk and limbs, accompanied by dystonic posturing of her upper limbs (video segment 1, Supplementary Video 1 in the online-only Data Supplement). Tone was increased in all limbs, with normal tendon reflexes and downgoing plantar responses.
Blood investigations including full blood count, renal, liver and thyroid function tests, electrolytes, ammonia, and vitamin B12 were normal. Syphilis and Human Immunodeficiency Virus serology were negative. Serum anti-thyroid peroxidase and thyroglobulin antibodies, connective tissue disease screen (antinuclear, anti-double stranded DNA and anti-cardiolipin antibodies, and lupus anticoagulant), paraneoplastic screen (anti-CV2, Yo, Ri, Hu, amphiphysin, and Ma antibodies), and tumor markers were negative. There were no acanthocytes in the peripheral blood film. Computed tomography of the thorax, abdomen and pelvis was unremarkable.
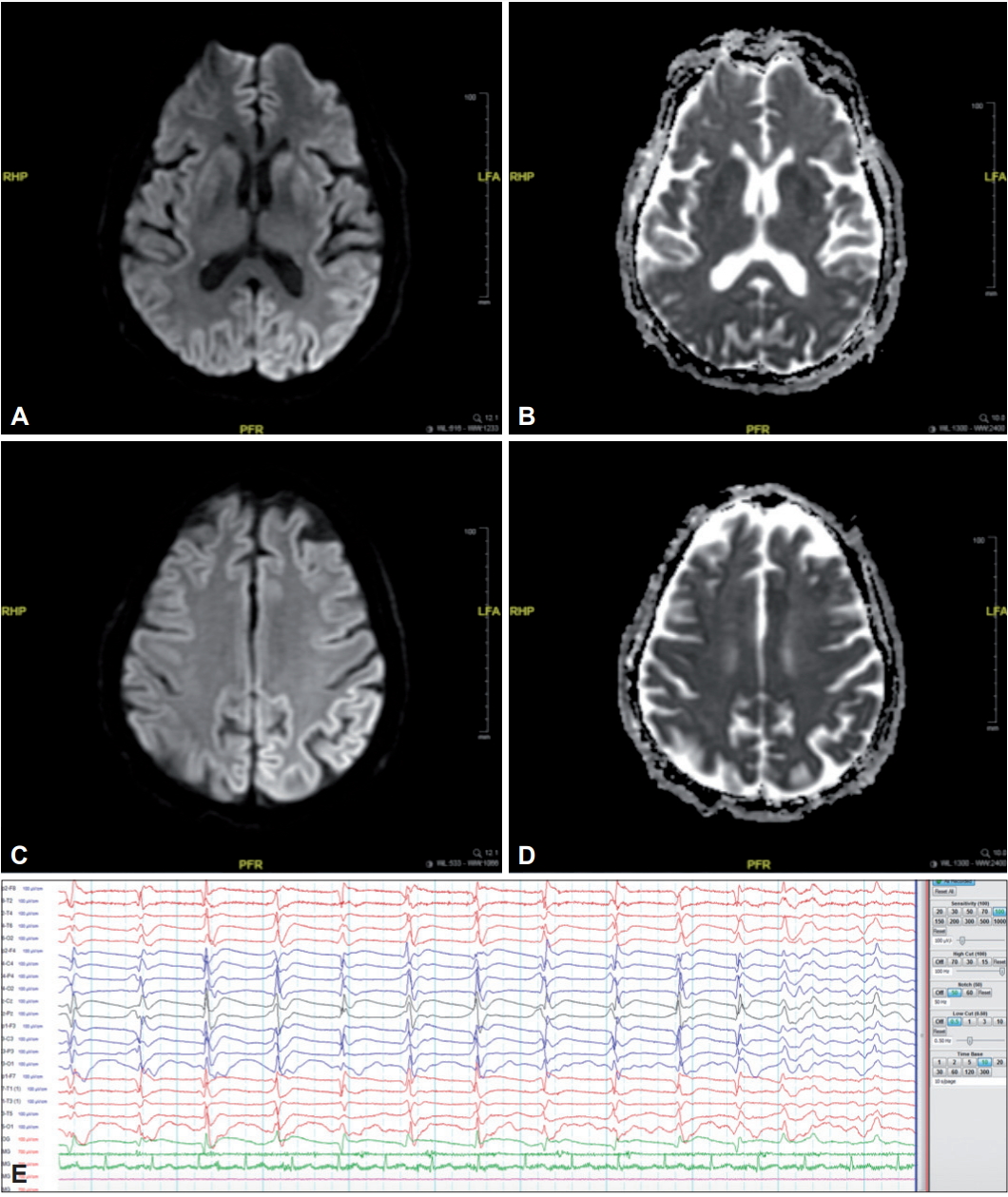
Brain magnetic resonance imaging (MRI) showed restricted diffusion in the caudate nucleus and putamen bilaterally with cortical ribboning in the occipito-parietal region bilaterally (Figure 1), without contrast enhancement. There was no significant brain atrophy or signal abnormalities on T1 or fluid-attenuated inversion recovery sequences. Initial electroencephalogram (EEG) showed diffuse polymorphic theta slowing with intermittent bilateral independent frontal sharp waves. Cerebrospinal fluid (CSF) analysis revealed normal opening pressure, cell counts and biochemistry. CSF autoimmune encephalitis screen (for anti-NMDAR, AMPAR-1, AMPAR-2, GABA-B, CASPR-2, and LGI-1 antibodies) was negative.
She was given a trial of intravenous methylprednisolone 1 g daily over five days, with no improvement. A repeat EEG showed a slower background rhythm with generalized periodic sharp-wave complexes (PSWCs) (Figure 1). A diagnosis of clinically probable sCJD was made based on the clinical history and progression, neuroimaging findings, evolving EEG changes and a subsequent positive CSF 14-3-3 protein result [3]. During her twelve-week hospital stay, she remained bedridden with no meaningful response. Her choreo-dystonic movements gradually diminished, while myoclonic jerks became increasingly frequent (video segment 2, Supplementary Video 1 in the onlineonly Data Supplement). She was eventually discharged home with community hospice support. Six months after symptom onset, she remained in a state of akinetic-mutism.
Although a definitive diagnosis of sCJD can only be made by histopathological examination, our patient more than fulfilled the minimum clinical criteria for probable sCJD based on clinical features, EEG showing PSWCs, CSF 14-3-3 positivity, and classical brain MRI abnormalities. In contrast, vCJD typically affects younger patients, shows the “pulvinar sign” on brain MRI and to our knowledge has not been reported in the South-East Asian region (the epidemic peaked in the late 1990s in the United Kingdom and has since tailed off). We believe that the other main differential diagnoses including infective encephalitis, autoimmune limbic encephalitis, toxic/metabolic syndromes, and non-convulsive status epilepticus were adequately excluded on the basis of the clinical features and extensive investigations. In this case of sCJD, we present an illustrative video showing an interesting evolution of movement disorders from predominant chorea-dystonia in the initial phase to akinetic mutism with myoclonus.
There are several single case reports of chorea in sCJD [2]. In a series of prospectively collected autopsy-proven cases, chorea was reported to be present in one out of 15 patients with sCJD [4]. Retrospective reviews of neuropathologically confirmed CJD cases revealed a 10–11% prevalence of chorea [1,5]; however, these series did not appear to distinguish between sCJD and vCJD. In other series, the authors categorized the movement disorders as “hyperkinesias” or “abnormal movements other than myoclonus,” but the prevalence of chorea was not specifically reported [3,6]; Brown et al. [6] (n = 230, neuropathologically verified CJD cases) specifically mentioned choreo-athetosis as a feature at illness onset in only one patient.
The movement disorders seen in CJD are thought to be a consequence of lesions in various structures including the striato-pallidal complex, the mesencephalon and the thalamus [5]. Chorea has been postulated to be due to dysfunction of the thalamus [2], a structure commonly involved in vCJD. The clinico-pathological features of CJD, particularly sCJD, are influenced by several factors including polymorphism of the prion protein gene (PRNP) involving methionine (Met) and valine (Val) at codon 129 and prion strain type (type 1 PrPSc or type 2 PrPSc) [7]. Myoclonus, PSWCs on EEG, and akinesia have been associated with Met-homozygosity, while ataxia has been associated with Val-homozygosity [1,7]. One case of sCJD with chorea was reported to be Val-homozygous [4]. In neuropathology studies, the first appearance of myoclonus in sCJD corresponds to spongiform changes in the cerebral neocortex with hypertrophic astrocytosis and ballooned neurons, while the akinetic mutism state represents an end-stage with extensive tissue rarefaction and neuron loss in the cerebral neocortex [7]. However, clinicopathological correlations for chorea in CJD have been inadequately described, and comprehensive analyses of PRNP, prion strain and neuropathology are needed to better understand these “atypical” sCJD cases.
In conclusion, although myoclonus and cerebellar ataxia are the classic movement disorders encountered in sCJD, we highlight here that chorea is a feature also compatible with the diagnosis. Awareness of this finding may potentially aid in the diagnostic workup of suspected sCJD cases.
Segment 1: Generalized chorea involving the face, trunk and limbs, accompanied by dystonic posturing of the upper limbs, two months after first symptom onset. Segment 2: During the patient’s 12-week hospital stay, the choreo-dystonic movements gradually diminished, while myoclonic jerks became increasingly frequent.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgments
The authors gratefully acknowledge the patient’s family for their consent and participation in this report, including publication of the video.
Figure 1.
Brain MRI showing restricted diffusion at both the caudate nucleus and putamen on the diffusion-weighted imaging (DWI) sequence (A) and apparent diffusion coefficient (ADC) map (B), with cortical ribboning (also with restricted diffusion) at the left occipito-parietal region on the DWI sequence (C) and ADC map (D). Electroencephalogram showing slow background rhythm with generalized periodic sharp-wave complexes (E).
REFERENCES
1. Edler J, Mollenhauer B, Heinemann U, Varges D, Werner C, Zerr I, et al. Movement disturbances in the differential diagnosis of Creutzfeldt-Jakob disease. Mov Disord 2009;24:350–356.ArticlePubMed
2. Maltête D, Guyant-Maréchal L, Mihout B, Hannequin D. Movement disorders and Creutzfeldt-Jakob disease: a review. Parkinsonism Relat Disord 2006;12:65–71.ArticlePubMed
3. Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–233.ArticlePubMed
4. Vital A, Fernagut PO, Canron MH, Joux J, Bezard E, Martin-Negrier ML, et al. The nigrostriatal pathway in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 2009;68:809–815.ArticlePubMed
5. Zochodne DW, Young GB, McLachlan RS, Gilbert JJ, Vinters HV, Kaufmann JC. Creutzfeldt-Jakob disease without periodic sharp wave complexes: a clinical, electroencephalographic, and pathologic study. Neurology 1988;38:1056–1060.ArticlePubMed
6. Brown P, Cathala F, Castaigne P, Gajdusek DC. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 1986;20:597–602.ArticlePubMed
7. Iwasaki Y. Creutzfeldt-Jakob disease. Neuropathology 2017;37:174–188.ArticlePubMed
Figure & Data
References
Citations
Citations to this article as recorded by
Atypical and early symptoms of sporadic Creutzfeldt – Jakob disease: case series and review of the literature Grammatiki Katsikaki, Ioannis E. Dagklis, Petros Angelopoulos, Dimitrios Ntantos, Angeliki Prevezianou, Sevasti Bostantjopoulou International Journal of Neuroscience.2021; 131(9): 927. CrossRef
Review of Hereditary and Acquired Rare Choreas Daniel Martinez-Ramirez, Ruth H. Walker, Mayela Rodríguez-Violante, Emilia M. Gatto Tremor and Other Hyperkinetic Movements.2020;[Epub] CrossRef
A Case of Creutzfeldt-Jakob Disease Presented as Rapid Progressive Parkinsonism Yoonah Park, Chan-Nyeong Lee Dementia and Neurocognitive Disorders.2019; 18(4): 152. CrossRef
 E-submission
E-submission
 , Tsun Haw Toh1, Soon Chai Low1, Si Lei Fong1, Kah Kian Chong1, Kee Wei Lee1, Khean Jin Goh1, Shen-Yang Lim1,2
, Tsun Haw Toh1, Soon Chai Low1, Si Lei Fong1, Kah Kian Chong1, Kee Wei Lee1, Khean Jin Goh1, Shen-Yang Lim1,2 

 PubReader
PubReader ePub Link
ePub Link Cite
Cite