ABSTRACT
-
Objective
- The survival of Huntington’s disease (HD) patients is reported to be 15–20 years. However, most studies on the survival of HD have been conducted in patients without genetic confirmation with the possible inclusion of non-HD patients, and all studies have been conducted in Western countries. The survival of patients with HD in East Asia, where its prevalence is 10–50-fold lower compared with Western populations, has not yet been reported.
-
Methods
- Forty-seven genetically confirmed Korean HD patients from independent families were included in this retrospective medical record review study.
-
Results
- The mean age at onset among the 47 patients was 46.1 ± 14.0 years. At the time of data collection, 25 patients had died, and these patients had a mean age at death of 57.8 ± 13.7 years. The Kaplan-Meier estimate of the median survival from onset in the 47 patients was 14.5 years (95% confidence interval: 12.3–16.6). None of the following factors were associated with the survival time in the univariate Cox regression analysis: gender, age at onset, normal CAG repeat size, mutant CAG repeat size, and the absence or presence of non-motor symptoms at onset.
-
Conclusion
- This is the first Asian study on survival in HD patients. Survival in Korean HD patients may be shorter than that reported for Western populations, or at least is in the lower range of expected survival. A larger longitudinal observation study is needed to confirm the results found in this study.
-
Keywords: Huntington’s disease; survival; CAG; Korea; Asia
Huntington’s disease (HD) is an autosomal-dominant progressive neurodegenerative disorder caused by CAG repeat expansions in the huntingtin gene. HD is characterized by progressive cognitive decline, psychiatric dysfunction, and movement disorder with a mean age of onset of 40 years [1]. There are great ethnic and geographic differences in the prevalence of HD. The prevalence of HD in Asian populations is 0.2–0.6 per 100,000 [2,3], whereas the prevalence among Western populations was determined to be 5.7 per 100,000 in a recent meta-analysis [3], and a prevalence greater than 10 per 100,000 has been reported in Canada and the UK [4,5]. The reason for the lower prevalence in Asian populations can be, at least partly, explained by the differences in the CAG repeat size, HTT haplotypes, and the CCG polymorphism in a region adjacent to the CAG repeat of the huntingtin gene [6-9], although underreporting cannot be completely excluded as another contributing factor.
The survival of HD patients is reported to be 15–20 years [1,10]. However, most studies on the survival of HD patients have been conducted in patients without genetic confirmation and with the possible inclusion of non-HD patients, and the results of those studies are inconsistent regarding factors influencing survival. Furthermore, all studies have been conducted in Western countries [11-15]. To our knowledge, survival in HD patients in Asia, where its prevalence is 10–50-fold lower compared with Western populations, has not been reported yet.
The aim of this study was to estimate the survival of HD patients in Korea and to examine the factors influencing survival.
MATERIALS & METHODS
- Forty-seven genetically confirmed HD patients from independent families who visited the neurology department of Seoul National University Hospital (SNUH) from 1994 to 2015 were included in this study. Thirty-six of the 47 (76.6%) patients were included in our previous report [16]. Demographic and clinical information was obtained by retrospective medical record review. Age at onset was defined as the age at which the patient developed motor symptoms or non-motor symptoms attributable to HD. Non-motor symptoms at onset were considered present if unequivocal cognitive decline or psychiatric symptoms developed before the onset of motor symptoms or simultaneously with motor symptoms. The vital status of all patients as of December 2015, as well as the date of death if deceased, was ascertained using data from the Korean National Statistics Office.
- Data were expressed as the mean ± SD. Correlations of the age at onset with the CAG repeat size were analyzed with Pearson’s correlation coefficients. Survival was analyzed by the Kaplan-Meier method, and the differences between genders were examined with the log-rank test. The effect of variables on survival was evaluated by Cox proportional hazards modelling. The following variables were evaluated in the Cox proportional hazards model: gender, age at onset, normal CAG repeat size, mutant CAG repeat size, and the absence or presence of non-motor symptoms at onset. All analyses were performed with SPSS 21.0 (SPSS Inc., Chicago, IL, USA). The level of statistical significance was set at p < 0.05. The study protocol was approved by the Seoul National University Hospital Institutional Review Board and conformed to the principles of the Declaration of Helsinki.
RESULTS
- The demographic, clinical, and genetic data are presented in Table 1. The year of symptom onset ranged from 1989 to 2011. Motor symptoms including chorea, ataxia, parkinsonism, and dystonia were the initial symptoms of 43 patients as a sole symptom (n = 20) or in combination with other non-motor symptoms including cognitive decline and psychiatric disturbances (n = 23), while four patients only developed non-motor symptoms as an initial symptom. There was no gender difference in the age at onset, symptoms at onset, and CAG repeat size in normal and mutant alleles. The CAG repeat size in the mutant allele showed an inverse correlation with the age at onset (r = -0.674, p < 0.001).
- At the time of data collection, 25 patients had died with a mean age at death of 57.8 ± 13.7 (range, 36–80) years (Table 1). The Kaplan-Meier estimate of median survival from onset in the 47 patients was 14.5 years [95% confidence interval (CI): 12.3–16.6] (Figure 1A). No difference in survival was observed between genders in the log-rank test (Figure 1B). In the univariate Cox regression analysis, none of the tested variables were associated with the survival time (Table 2).
DISCUSSION
- In this first Asian study on survival in HD patients, the median survival from onset was 14.5 years. Although a direct comparison is not possible, it appears that the mean survival in our study is shorter that that reported by Rinaldi et al. [14] (20 years, 95% CI: 18.3–21.7). In a study by Pekmezovic et al. [13], the cumulative probabilities of survival in 5, 10, 15, and 20-year periods calculated by the life table method were 91, 63, 10, and 5%, respectively, compared to 88, 49, 26, and 5% in our patients, respectively. Taking into account the studies without a genetic diagnosis which report a survival of 15–20 years as well [11,12,15], the survival in our patients appears to be shorter than that reported for Western populations, or at least is in the lower range of the estimated survival. A couple of reasons may explain the shorter survival observed in our patients. First, the mean age at onset in our patients was higher by approximately 5 years compared with studies conducted in Western populations, despite a similar mutant CAG repeat size. Specifically, when compared with the results of Rinaldi et al. [14], the mean age at onset was higher by 6 years, despite the fact that the mean age at death was similar. This later onset with a similar age at death may explain the shorter survival in our patients. However, it is not clear whether HD patients in Korea really have a later age of onset or whether this finding is due to a delay in diagnosis. Of note, a recent study from Korea showed an onset age of 44.16 ± 14.08 years [2], which is similar to our result. Intriguingly, the age at onset of HD patients in Chinese populations was reported to be in the mid-30s [17,18]. It will be interesting to evaluate survival of HD patients in these populations. Second, differences in the HTT haplotypes and CCG polymorphisms, which are related to the differences in the HD prevalence between populations [6-9], might have a role in the difference in survival, although there is no evidence yet that these genetic factors have any effect on the progression or survival of HD patients. It cannot be excluded that yet unknown genetic factors may have an effect on the survival of HD patients. Third, survival among patients with progressive neurodegenerative disorders like HD is influenced not only by the disease itself but also by other sociocultural factors and the prevalence of other disorders affecting the elderly in that population.
- In our study, none of the tested variables were associated with the survival time. The lack of an association between the mutant CAG repeat size with survival time is in contrast with recent studies on genetically confirmed HD patients [13,14]. No correlation of age at onset with the survival time is also in contrast with the findings of previous studies. Older age at onset has repeatedly been associated with shorter survival [11,12,14,15]. However, in a recent study by Pekmezovic et al. [13] on 112 Serbian patients with genetically confirmed HD, older age at onset was associated with longer survival. The reason for these discrepancies might be due to the differences in the distribution of age at onset, especially the proportion of juvenile-onset HD and late-onset HD. Juvenile onset HD has been associated with a shorter survival in some studies [11,13], and Pekmezovic et al. [13] reported a 10-year survival rate of 0% in three juvenile-onset cases. In our study, one patient had juvenile-onset HD (18 years old). This male patient was still alive at the time of data analysis, with a disease duration of 15 years. In contrast to many studies showing longer survival in female patients [11-13,15], our study showed no gender difference. A recent study by Rinaldi et al. [14] on 135 Italian patients with genetically confirmed HD also reported no gender difference in survival when controlling for age at onset and the mutant CAG repeat size.
- This study has several limitations. First, the number of patients is small, which was due to the low prevalence of HD in Korea. Second, our use of a retrospective design with medical records limited the accuracy of the clinical history data. Third, selection bias cannot be excluded because SNUH is a tertiary referral hospital. Forth, the age at onset in this study was defined as the age at which the patient developed motor symptoms, cognitive decline, or psychiatric symptoms, rather than the age at motor onset, which is more conventionally used. Interpretation of survival in this study requires caution in this regard. However, recent studies on the survival of HD patients have used the same definition of age at onset as that used in our study. If we had defined the age at onset as the age at motor onset, the mean age at onset would have been higher, and survival would have been shorter.
- A clear understanding of survival in HD patients and the factors influencing survival is important for patient care and may help to identify factors related to disease progression in HD patients. Current, ongoing, large longitudinal observational studies will provide a better understanding in this regard.
Notes
-
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgments
- The authors would like to thank Dr. Caroline H. Williams-Gray (John Van Geest Centre for Brain Repair, University of Cambridge) for her advice on preparing the manuscript.
Figure 1.A: Kaplan-Meier survival curve for the 47 patients. The dotted line indicates median survival. B: Kaplan-Meier survival curves for men (solid line) and women (dotted line).
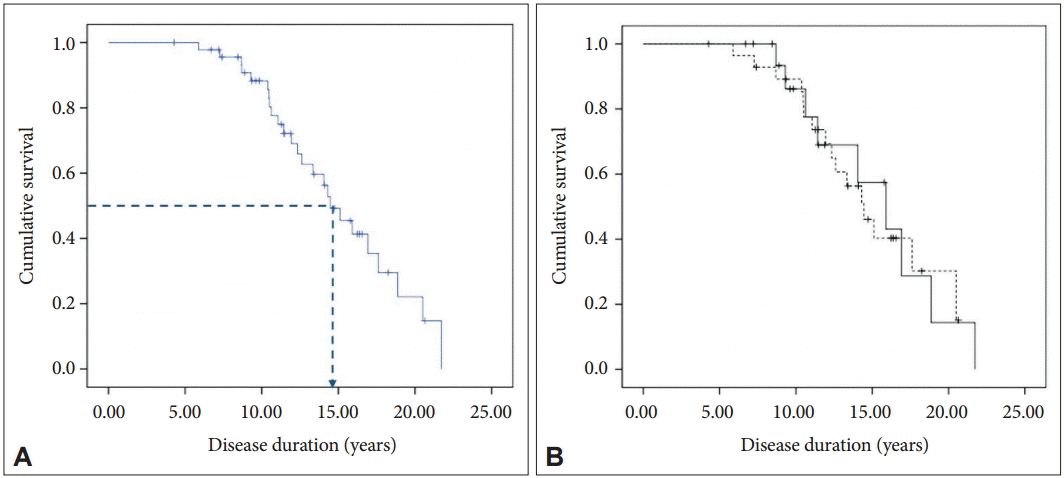
Table 1.Demographic, clinical, and genetic characteristics
|
Characteristics |
Total (n = 47) |
Deceased (n = 25) |
Non-deceased (n = 22) |
|
Age at onset |
46.1 ± 14.0 (range, 18–71) |
44.8 ± 14.0 (range, 22–70) |
47.8 ± 14.3 (range, 18–70) |
|
Age at death |
|
57.8 ± 13.7 (range, 36–80) |
|
|
Duration at death (yr) |
|
13.0 ± 4.0 |
|
|
Male:female |
18:29 |
9:16 |
9:13 |
|
Family history |
|
|
|
|
Present |
33 |
17 |
16 |
|
Absent |
8 |
5 |
3 |
|
Unknown |
6 |
3 |
3 |
|
Symptom at onset |
|
|
|
|
Involuntary movement |
43 |
24 |
19 |
|
Cognitive decline |
20 |
12 |
8 |
|
Psychiatric disturbance |
11 |
4 |
7 |
|
Mutant CAG repeat length |
44.9 ± 4.5 (range, 39–58) |
46.1 ± 4.4 (range, 40–56) |
43.6 ± 4.4 (range, 39–58) |
|
Wild type CAG repeat length |
18.5 ± 2.1 (range, 15–24) |
19.1 ± 2.4 (range, 15–24) |
17.7 ± 1.5 (range, 15–20) |
Table 2.Results of univariate Cox proportional hazards analysis
|
Baseline variable |
Unadjusted hazard ratio (95% CI) |
p-value |
|
Gender |
0.957 (0.408–2.248) |
0.920 |
|
Age at onset |
1.027 (0.994–1.061) |
0.112 |
|
Normal CAG repeat size |
1.077 (0.882–1.317) |
0.466 |
|
Mutant CAG repeat size |
1.044 (0.961–1.134) |
0.304 |
|
Non-motor symptom at onset |
0.917 (0.400–2.103) |
0.839 |
REFERENCES
- 1. Walker FO. Huntington’s disease. Lancet 2007;369:218–228.ArticlePubMed
- 2. Kim HS, Lyoo CH, Lee PH, Kim SJ, Park MY, Ma HI, et al. Current status of Huntington’s disease in Korea: a Nationwide Survey and National Registry Analysis. J Mov Disord 2015;8:14–20.ArticlePubMedPMC
- 3. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 2012;27:1083–1091.ArticlePubMed
- 4. Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry 2013;84:1156–1160.ArticlePubMedPMC
- 5. Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 2014;29:105–114.ArticlePubMed
- 6. Pêcheux C, Mouret JF, Dürr A, Agid Y, Feingold J, Brice A, et al. Sequence analysis of the CCG polymorphic region adjacent to the CAG triplet repeat of the HD gene in normal and HD chromosomes. J Med Genet 1995;32:399–400.ArticlePubMedPMC
- 7. Morovvati S, Nakagawa M, Osame M, Karami A. Analysis of CCG repeats in Huntingtin gene among HD patients and normal populations in Japan. Arch Med Res 2008;39:131–133.ArticlePubMed
- 8. Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet 1994;3:2103–2114.ArticlePubMedPDF
- 9. Warby SC, Visscher H, Collins JA, Doty CN, Carter C, Butland SL, et al. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet 2011;19:561–566.ArticlePubMedPMC
- 10. Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98.ArticlePubMed
- 11. Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry 1999;66:52–56.ArticlePubMedPMC
- 12. Newcombe RG, Walker DA, Harper PS. Factors influencing age at onset and duration of survival in Huntington’s chorea. Ann Hum Genet 1981;45(Pt 4):387–396.ArticlePubMed
- 13. Pekmezovic T, Svetel M, Maric J, Dujmovic-Basuroski I, Dragasevic N, Keckarevic M, et al. Survival of Huntington’s disease patients in Serbia: longer survival in female patients. Eur J Epidemiol 2007;22:523–526.ArticlePubMed
- 14. Rinaldi C, Salvatore E, Giordano I, De Matteis S, Tucci T, Cinzia VR, et al. Predictors of survival in a Huntington’s disease population from southern Italy. Can J Neurol Sci 2012;39:48–51.ArticlePubMed
- 15. Roos RA, Hermans J, Vegter-van der Vlis M, van Ommen GJ, Bruyn GW. Duration of illness in Huntington’s disease is not related to age at onset. J Neurol Neurosurg Psychiatry 1993;56:98–100.ArticlePubMedPMC
- 16. Shin CW, Choi YJ, Kim M, Jeon BS. Preliminary analysis of Huntington’s Disease in South Korea. J Huntingtons Dis 2013;2:83–87.ArticlePubMed
- 17. Guo X, Zhang S, Burgunder JM, Shang HF. Clinical features of Huntington disease in 243 Chinese patients. Neural Regen Res 2010;5:102–107.
- 18. Leung CM, Chan YW, Chang CM, Yu YL, Chen CN. Huntington’s disease in Chinese: a hypothesis of its origin. J Neurol Neurosurg Psychiatry 1992;55:681–684.ArticlePubMedPMC
Citations
Citations to this article as recorded by

- Analysis of HTT CAG repeat expansion among healthy individuals and patients with chorea in Korea
Ryul Kim, Moon-Woo Seong, Bumjo Oh, Ho Seop Shin, Jee-Soo Lee, Sangmin Park, Mihee Jang, Beomseok Jeon, Han-Joon Kim, Jee-Young Lee
Parkinsonism & Related Disorders.2024; 118: 105930. CrossRef - Clinical and Genetic Characteristics Associated With
Survival Outcome in Late-Onset Huntington’s Disease
in South Korea
Yun Su Hwang, Sungyang Jo, Gu-Hwan Kim, Jee-Young Lee, Ho-Sung Ryu, Eungseok Oh, Seung-Hwan Lee, Young Seo Kim, Sun Ju Chung
Journal of Clinical Neurology.2024;[Epub] CrossRef - Increased 10-Year Prevalence of Huntington’s Disease in South Korea: An Analysis of Medical Expenditure Through the National Healthcare System
Chan Young Lee, Jun-soo Ro, Hyemin Jung, Manho Kim, Beomseok Jeon, Jee-Young Lee
Journal of Clinical Neurology.2023; 19(2): 147. CrossRef - Clustering and prediction of disease progression trajectories in Huntington's disease: An analysis of Enroll-HD data using a machine learning approach
Jinnie Ko, Hannah Furby, Xiaoye Ma, Jeffrey D. Long, Xiao-Yu Lu, Diana Slowiejko, Rita Gandhy
Frontiers in Neurology.2023;[Epub] CrossRef - Survival in Huntington’s disease and other young‐onset dementias
Samantha M. Loi, Paraskevi Tsoukra, Emily Sun, Zhibin Chen, Pierre Wibawa, Maria di Biase, Sarah Farrand, Dhamidhu Eratne, Wendy Kelso, Andrew Evans, Mark Walterfang, Dennis Velakoulis
International Journal of Geriatric Psychiatry.2023;[Epub] CrossRef - Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study
Roberto D. V. S. Morais, Marina Sogorb-González, Citlali Bar, Nikki C. Timmer, M. Leontien Van der Bent, Morgane Wartel, Astrid Vallès
Cells.2022; 11(17): 2748. CrossRef - Huntington's disease: Mortality and risk factors in an Australian cohort
Emily Sun, Matthew Kang, Pierre Wibawa, Vivian Tsoukra, Zhibin Chen, Sarah Farrand, Dhamidhu Eratne, Wendy Kelso, Andrew Evans, Mark Walterfang, Dennis Velakoulis, Samantha M. Loi
Journal of the Neurological Sciences.2022; 442: 120437. CrossRef - Huntington’s disease in Turkey: genetic counseling, clinical features, and outcome
Yesim Sucullu Karadag, Busranur Erozan Cavdarli, Rabia Nazik Yuksel
Neurological Research.2021; 43(5): 381. CrossRef - Validation of diagnostic codes and epidemiologic trends of Huntington disease: a population-based study in Navarre, Spain
Esther Vicente, Ainara Ruiz de Sabando, Fermín García, Itziar Gastón, Eva Ardanaz, María A. Ramos-Arroyo
Orphanet Journal of Rare Diseases.2021;[Epub] CrossRef - Epidemiology of Huntington disease in Cyprus: A 20‐year retrospective study
C.A. Demetriou, A. Heraclides, C. Salafori, G.A. Tanteles, K. Christodoulou, Y. Christou, E. Zamba‐Papanicolaou
Clinical Genetics.2018; 93(3): 656. CrossRef - Population-specific genetic modification of Huntington's disease in Venezuela
Michael J. Chao, Kyung-Hee Kim, Jun Wan Shin, Diane Lucente, Vanessa C. Wheeler, Hong Li, Jared C. Roach, Leroy Hood, Nancy S. Wexler, Laura B. Jardim, Peter Holmans, Lesley Jones, Michael Orth, Seung Kwak, Marcy E. MacDonald, James F. Gusella, Jong-Min L
PLOS Genetics.2018; 14(5): e1007274. CrossRef
 E-submission
E-submission

 PubReader
PubReader ePub Link
ePub Link Cite
Cite
