E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 17(1); 2024 > Article
-
Review Article
Ultrastructures of α-Synuclein Filaments in Synucleinopathy Brains and Experimental Models -
Airi Tarutani

 , Masato Hasegawa
, Masato Hasegawa -
Journal of Movement Disorders 2024;17(1):15-29.
DOI: https://doi.org/10.14802/jmd.23213
Published online: November 22, 2023
Department of Brain and Neurosciences, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
- Corresponding author: Airi Tarutani, PhD Department of Brain and Neurosciences, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan / Tel: +81-3-6834-2349 / Fax: +81-3-6834-2349 / E-mail: tarutani-ar@igakuken.or.jp
- Corresponding author: Masato Hasegawa, PhD Department of Brain and Neurosciences, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan / Tel: +81-3-6834-2349 / Fax: +81-3-6834-2349 / E-mail: hasegawa-ms@igakuken.or.jp
Copyright © 2024 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,386 Views
- 156 Download
- ABSTRACT
- INTRODUCTION
- α-SYN FILAMENTS IN SYNUCLEINOPATHY BRAINS
- α-SYN FILAMENTS IN IN VITRO MODELS
- α-SYN FILAMENTS IN CELLULAR MODELS
- α-SYN FILAMENTS IN ANIMAL MODELS
- IMPLICATIONS OF CONFORMATIONAL DIFFERENCES BETWEEN PATIENT- AND EXPERIMENTAL MODEL-DERIVED α-SYN FILAMENTS
- CONCLUSIONS
- Notes
- Acknowledgments
- REFERENCES
ABSTRACT
- Intracellular α-synuclein (α-syn) inclusions are a neuropathological hallmark of Lewy body disease (LBD) and multiple system atrophy (MSA), both of which are termed synucleinopathies. LBD is defined by Lewy bodies and Lewy neurites in neurons, while MSA displays glial cytoplasmic inclusions in oligodendrocytes. Pathological α-syn adopts an ordered filamentous structure with a 5–10 nm filament diameter, and this conformational change has been suggested to be involved in the disease onset and progression. Synucleinopathies also exhibit characteristic ultrastructural and biochemical properties of α-syn filaments, and α-syn strains with distinct conformations have been identified. Numerous experimental studies have supported the idea that pathological α-syn self-amplifies and spreads throughout the brain, during which processes the conformation of α-syn filaments may drive the disease specificity. In this review, we summarize the ultrastructural features and heterogeneity of α-syn filaments in the brains of patients with synucleinopathy and in experimental models of seeded α-syn aggregation.
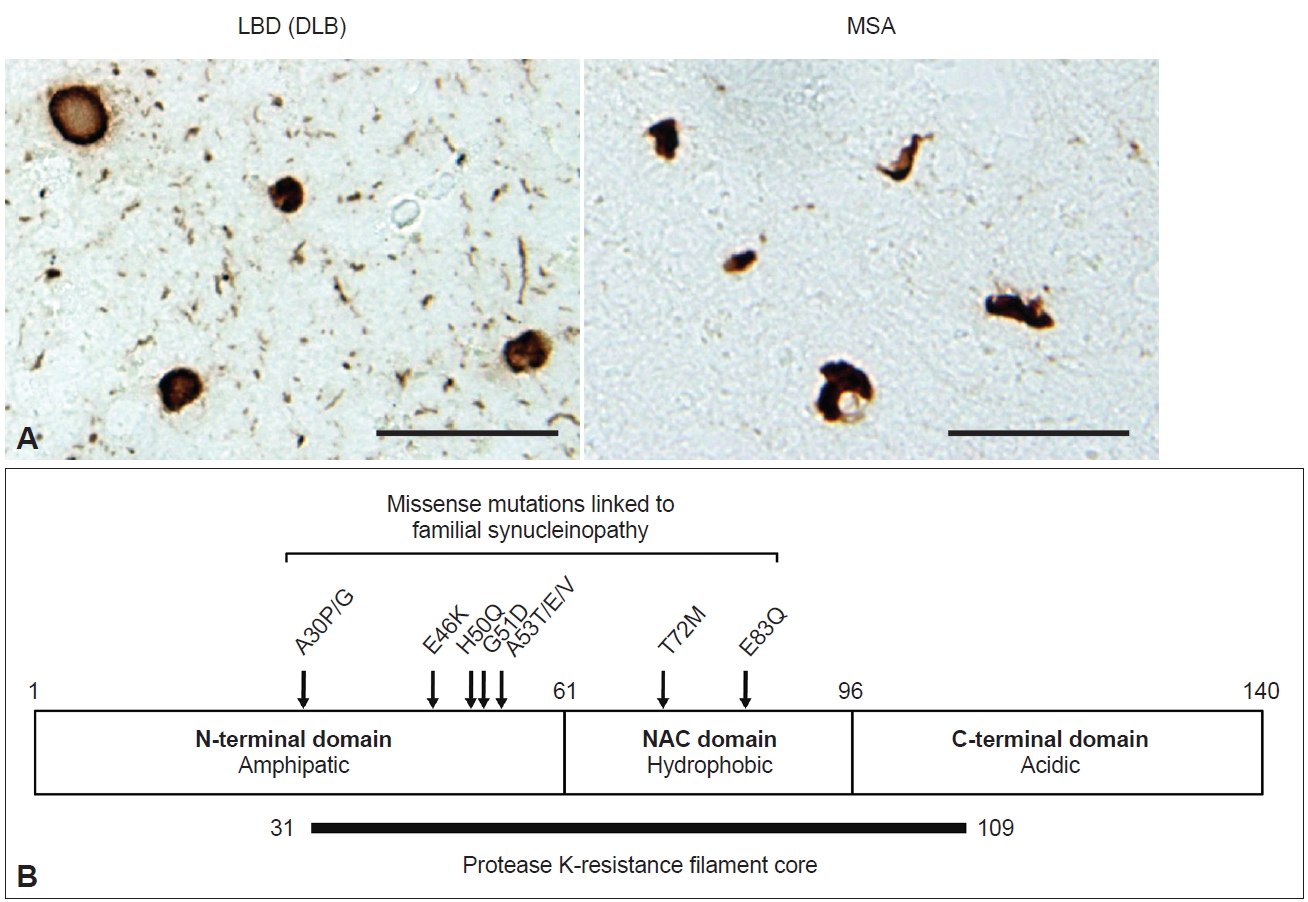
- Synucleinopathies are a subset of neurodegenerative diseases characterized by inclusions in which α-synuclein (α-syn) is a major component. In Lewy body disease (LBD), including Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB), Lewy bodies (LBs) characterized by an eosinophilic core and a surrounding clear halo are observed in neurons, and these structures are commonly described as brainstem-type LBs (Figure 1A) [1-3]. Cortical-type LBs lacking a distinctive core and halo are also observed in LBD, as well as Lewy neurites (LNs) in axons and dendrites (Figure 1A) [1,4]. In 1997, the A53T mutation in the SNCA gene encoding α-syn was reported to cause familial PD in Italian and Greek families [5]. This finding led to the identification of α-syn as a major component of LBs [6,7]. In 1998, it was further established that α-syn composes glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA) (Figure 1A) [8]. MSA is divided into two types based on motor phenotype: parkinsonian type (MSA-P), with predominant extrapyramidal symptoms, and cerebellar type (MSA-C), characterized by cerebellar dysfunction [9]. Currently [10], missense mutations (A30P, A30G, E46K, H50Q, G51D, A53T, A53E, A53V, T72M, E83Q) and multiplication (duplication, triplication) of the SNCA gene have been reported to be linked to the onset of synucleinopathy (Figure 1B) [5,10-20]. The above discoveries paved the way for the detailed characterization of α-syn-related pathogenesis both in vitro and in vivo.
- α-syn is a small protein consisting of 140 amino acids, with an amphipathic N-terminal domain (1–60 residues), a non-β amyloid component (NAC) domain (61–95 residues) and an acidic C-terminal domain (96–140 residues) (Figure 1B). The N-terminal domain adopts an α-helical structure, and seven 11-amino-acid imperfect repeats containing the KTEGV motif are found in the N-terminal and NAC domains [21,22]. The NAC domain, originally identified as peptides X and Y in the sodium dodecyl sulfate (SDS)-insoluble fraction prepared from Alzheimer’s disease (AD) brains [23], is hydrophobic and responsible for the amyloidogenic property of α-syn [24]. The C-terminal region is negatively charged, and its truncation (121–140 residues) enhances the cytotoxicity and aggregation of α-syn [25-27]. α-syn is highly expressed in neurons, accounting for approximately 1% of total proteins expressed in the brain, and is localized to presynaptic terminals [28]. Although its physiological function is uncertain, studies of α-syn-knockout mice indicate that α-syn is involved in dopamine release [29], and roles in the regulation of neurotransmitter release and synaptic plasticity through chaperoning SNARE complex assembly and clustering synaptic vesicles have been proposed [30-32]. While α-syn also has a potential role in the regulation of lipid metabolism [33-35], mutations in related genes, such as GBA1 and VSP35, are the most plausible genetic risk factors for LBD onset [36,37]. Under physiological conditions, α-syn is categorized as a natively unfolded protein and is highly water soluble [38]. In contrast, pathological α-syn extracted from synucleinopathy brains is conformationally converted into β-sheet-rich amyloid-like filaments, which are insoluble in detergents such as sarkosyl, SDS and Triton X-100 and exhibit resistance to proteases [39-43]. These properties are akin to those of the infectious prion protein that causes prion disease [44]. Protease K (PK) treatment of recombinant α-syn filaments revealed that residues 31–109 form the filament core (Figure 1B) [42]. Most missense mutations are located in this region, suggesting that structural changes in normal α-syn cause rapid aggregation and result in early disease onset. Phosphorylation at S129 (pS129) is detected at > 90% frequency in synucleinopathy brains [45] and has been widely used as a marker to distinguish normal α-syn from pathological α-syn in both clinical and research fields. In addition to pS129, phosphorylation at other residues, such as Y39, T59, T64, T72, T81, Y125, Y133, and Y136, ubiquitination at K6, K12, K23, K60, and K80, N-terminal acetylation, O-GlcNAcylation, nitration and C-terminal truncation have also been reported in pathological α-syn [27,46-49].
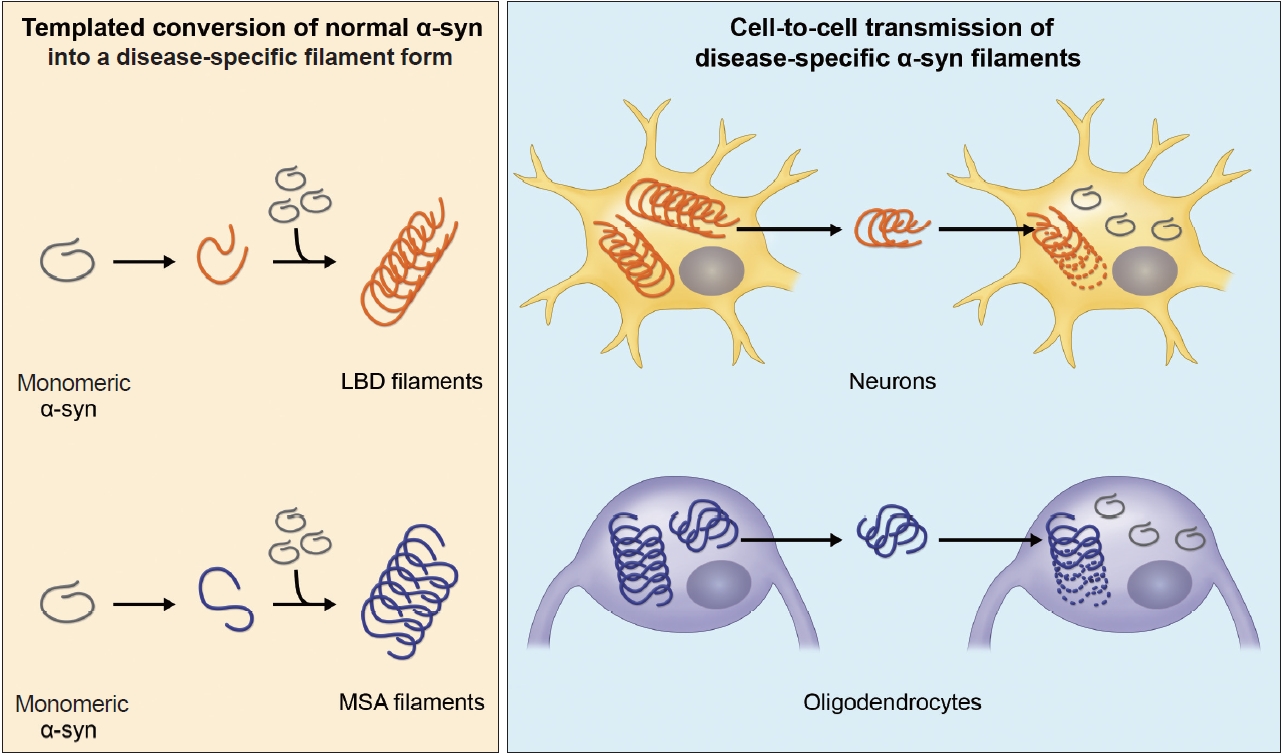
- Furthermore, α-syn filaments extracted from synucleinopathy brains and recombinant α-syn filaments have been demonstrated to work as “seeds” to trigger α-syn aggregation and pathology formation in in vitro and cellular systems and in animals, suggesting that the disease progression in LBD and MSA is based on prion-like amplification and spreading of pathological α-syn [50-52]. Disease-specific α-syn filaments recruit normal α-syn into a filament form in a templated manner and self-amplify (Figure 2). The amplified α-syn filaments then transmit from cell to cell and spread throughout the brain along neuronal networks (Figure 2). In these processes, disease specificity is thought to be maintained through inheritance of the conformation of the original seeds. Although the molecular mechanisms remain unclear, experimental studies have supported the idea that this prion-like hypothesis can explain the stereotypical expansion of α-syn pathology in LBD brains [53,54]. The observations of Lewy pathology in fetal dopamine neurons 10–24 years after transplantation into PD patients are also consistent with cell-to-cell transmission of pathological α-syn [55-57]. In recent years, cryo-electron microscopy (EM) structural analysis of patient-derived α-syn filaments has substantially advanced the understanding of the structural heterogeneity of the inclusions in synucleinopathies [48,58,59]. This review describes the characteristics of α-syn filaments accumulated in synucleinopathy brains and formed in experimental seeded aggregation models.
INTRODUCTION
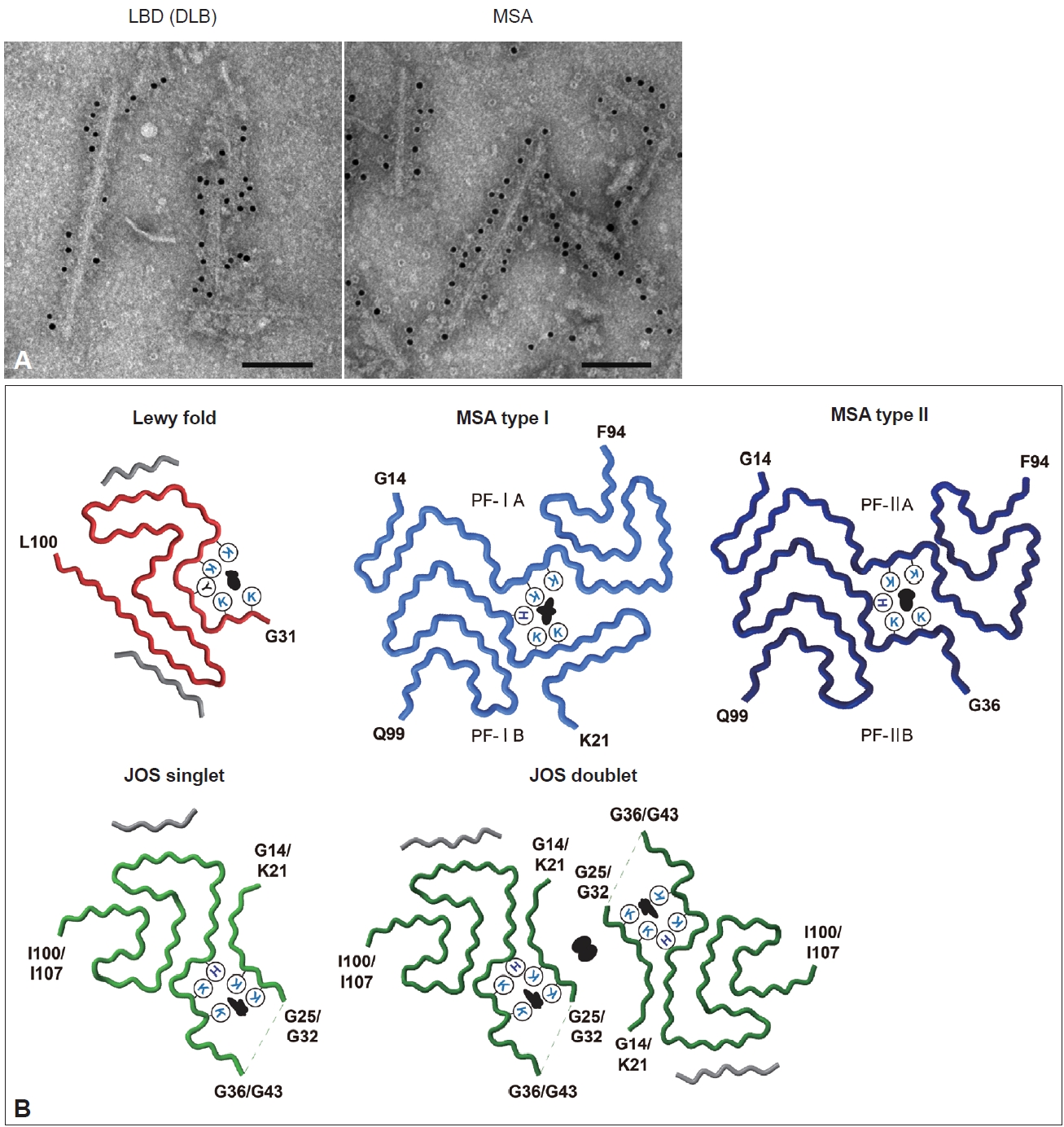
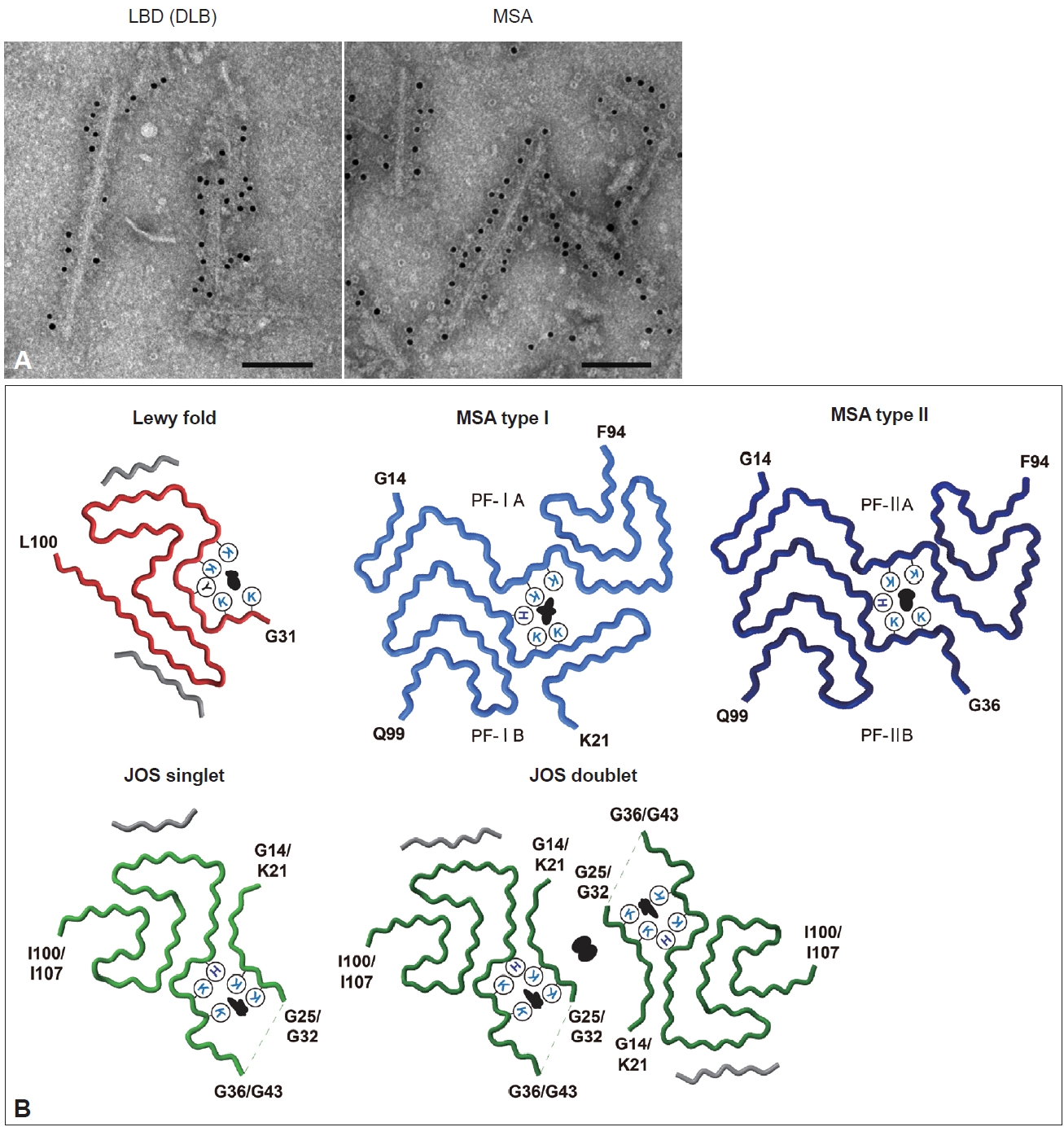
- Duffy and Tennyson [60] first reported in 1965 that LBs observed in PD are composed of filamentous structures. EM of LBD brain sections revealed that abundant 7–20 nm straight filaments and their bundles fill LBs, and are distinct from neurofilaments having side arms [60,61]. Furthermore, brainstem-type LBs form a dense central core with radiating filaments, whereas the filaments in cortical-type LBs are less organized and more diffuse [4]. Besides the filaments, which later turned out to contain α-syn, the presence of lipids, membrane fragments, globular materials, and partial cytoplasmic organelles in LBs was also described [60,62]. Recently, stimulated emission depletion-based superresolution microscopy demonstrated the high occupancy of lipids and organelles in Lewy pathology [63]. More than 100 proteins, including proteins related to proteolytic systems, oxidative stress, cell signaling and protein folding, have been reported to colocalize with α-syn in LBs [64-66]. GCIs in MSA brain sections are constructed of filaments approximately 25 nm in diameter, which are not tightly associated with the cell membranes [67,68]. α-syn filaments accumulated in synucleinopathy brains can be extracted by fractionation using detergents. Abundant filaments labeled with α-syn antibodies are observed in detergent-insoluble fractions [7]. The ultrastructures of the extracted filaments resemble those observed in the brain sections. LBD-derived α-syn filaments (LBD-α-syn) are observed as linear structures 5–10 nm in diameter, whereas MSA-derived α-syn filaments (MSA-α-syn) have the same diameter but exhibit a twist with 80–100 nm periodicity (Figure 3A) [69-71]. The C-terminal α-syn antibody labels the entirety of both α-syn filaments, whereas the N-terminal α-syn antibody recognizes only one side of the filament ends, suggesting that α-syn filaments have a polar structure [69,70,72]. The ultrastructural differences between LBD-α-syn and MSA-α-syn are also demonstrated by protease digestion of the nonfilament core region. PK treatment shows disease-specific PK-resistant α-syn banding patterns, indicating that LBD-α-syn and MSA-α-syn adopt distinct filament core structures [73]. Furthermore, MSA-α-syn exhibits higher SDS solubility than LBD-α-syn [40,74].
- In the last few years, structural polymorphisms of patient-derived α-syn filaments have been demonstrated at the atomic level by single-particle analysis using cryo-EM [48,58,59]. Two types of asymmetrical structures of α-syn filaments extracted from the putamen of five MSA cases, including MSA-P and MSA-C, were reported in 2020 (Figure 3B) [48]. These Type I and Type II filaments were each composed of two protofilaments with distinct filament cores. Protofilaments of Type I consist of G14-F94 (PF-IA) and K21-Q99 (PF-IB); the N-terminal region of PF-IA has an elongated one-layer L-shaped motif that is absent in PF-IB, and the C-terminal regions of both protofilaments show a compact three-layer L-shaped motif but with different packing. Type II filaments are made of protofilaments comprising G14-F94 (PF-IIA) and G36-Q99 (PF-IIB), which differ from those of Type I filaments. PF-IA and PF-IIA or PF-IB and PF-IIB show similar core structures, but PF-IIA contains a more extended cavity in the C-terminal region, and PF-IIB has a smaller N-terminal arm. Two types of substructures were identified at A78-Q99 in PF-IIB. In addition, a negatively charged additional density surrounded by K43, K45 and H50, which is distinguished from α-syn, is present between the two protofilaments of Type I and Type II. This suggests that some unidentified nonproteinaceous molecules are involved in the packing of the two protofilaments. The ratio of Type I to Type II in the putamen varied from case to case, and this variation was also observed in different brain regions of a single MSA case. Although the disease duration and the presence of neuronal cytoplasmic inclusions potentially alter the ratio of Type I to Type II, further investigation is needed to clarify what factors, such as brain regions, cell types, and other cellular environmental factors, influence this issue. In 2022, α-syn filaments extracted from the cingulate cortex or frontal cortex of six LBD cases, including PD, PDD, and DLB, were reported to show a common protofilament core structure termed the Lewy fold (Figure 3B) [58]. Most LBD-derived α-syn filaments are ultrastructurally straight, but the Lewy fold was determined from the twisted parts seen in approximately 25% of filaments. The Lewy fold consists of G31-L100, and nine β-strands form a three-layered core structure. Two islands with unknown amino acid sequences are packed in the Lewy fold. An additional density surrounded by K32, K34, Y39, K43 and K45, resembling in size those found in MSA, was also identified. Cryo-EM analysis of α-syn filaments derived from MSA and LBD brains showed that α-syn filaments with one or multiple common folds form disease-characteristic α-syn pathology, indicating that synucleinopathy can be defined in terms of α-syn filament structure. Since peripheral α-syn pathology is prominent in patients with LBD [75-78], it will also be important to determine the atomic structure of α-syn filaments derived from peripheral tissues. More recently, the juvenile-onset synucleinopathy (JOS) fold, which differs from the MSA folds and the Lewy fold, was identified in α-syn filaments extracted from one JOS case that developed DLB at 13 years of age (Figure 3B) [59,79]. Genetic analysis found a 7-amino-acid insertion (MAAAEKT) after T22, resulting from a 21-nucleotide duplication in one of the SNCA alleles. Immunoblot analysis of the detergent-insoluble fraction of this case detected bands of both wild-type (WT) α-syn (15 kDa) and insertion mutant α-syn comprising 147 amino acids (16 kDa). The JOS fold consists of G36-I100 in WT α-syn or G43-I107 in insertion mutant α-syn (the main core is not affected by the insertion) and two disconnected islands. Protofilaments and C2 symmetrically packed doublets were identified at a ratio of approximately 4:1. Partial similarity with the substructure common to Type I and Type II in MSA is found in the main core of the JOS fold. One island packed at the N-terminus of the main core appears to consist of G14-G25 in WT α-syn or K21-G32 in insertion mutant α-syn, and this island is probably essential for the JOS doublet structure. An additional density surrounded by K21/K28, K23/K30, K43/K50, K45/K52, and H50/H57 was present between this island and the main core. WT α-syn expressed in this JOS case did not adopt the Lewy fold, and WT and insertion mutant α-syn filaments showed a common fold. This may imply that insertion mutant α-syn first formed the JOS fold, then worked as a template, and recruited WT α-syn into the JOS fold form. Cryo-EM structures of patient-derived α-syn filaments provide evidence not only for the presence of α-syn strains in synucleinopathy, but also for templated amplification of disease-specific α-syn filaments in human brains.
α-SYN FILAMENTS IN SYNUCLEINOPATHY BRAINS
- Experimental seeded α-syn aggregation has been demonstrated in vitro and in cultured cells, as well as in primary cultures and in animals. Recombinant α-syn purified from Escherichia coli forms amyloid-like filaments 5–10 nm in diameter after shaking at 37°C for several days. Mutant and truncated α-syn filaments exhibit different ultrastructural and biochemical properties from WT filaments. Most α-syn mutants have been reported to show a faster increase in thioflavin fluorescence intensity compared to WT, though A30P and G51D α-syn show a slower increase [80-85]. A30P α-syn filaments are more fragile than WT filaments, and seeded A30P filaments generated using WT monomers are also fragile [82]. C-terminally truncated α-syn forms shorter, more twisted filaments, and filament formation is relatively rapid compared to that of full-length α-syn [86-88]. These various types of recombinant α-syn filaments exhibit distinct protease-resistant α-syn banding patterns [82], supporting the idea that conformational differences give rise to different prion-like properties. In addition, recombinant α-syn filament strains can be formed from identical α-syn monomers, depending on the buffer composition, the presence of cofactors, and other conditions, such as shaking and temperature. The formation of ribbon-like or straight α-syn filaments is induced in the presence or absence of inorganic salts, and these strains exhibit different aggregation kinetics and interactions with the 26S proteasome [89,90]. Glycosaminoglycans such as heparin, metal ions and lipid vesicles also promote α-syn aggregation, leading to structural diversity [91-93]. The structures of recombinant WT, mutant, and truncated α-syn filaments have been determined by cryo-EM analysis [94-105]. Recombinant α-syn filaments have no structural identity at the atomic level with patient-derived α-syn filaments and show variable filament cores according to mutations and in vitro conditions. However, the substructures of patient-derived filaments and several types of recombinant filaments share similarities. In particular, V48-T92 containing the NAC domain tends to adopt a three-layer L-shaped structure, which may be due to a smaller nonfilament core region.
- Additionally, amplification of α-syn contained in tissues and body fluids from patients with synucleinopathy has been utilized to develop the in vitro seed amplification assay (SAA), also known as PMCA or RT-QuIC [106,107]. Recombinant α-syn monomers and patient-derived samples are mixed and shaken at constant intervals during incubation. In general, different kinetics of thioflavin fluorescence intensities are seen in LBD and MSA [108,109], which may represent distinct bindings of thioflavin to the SAA product. Although SAA is undoubtedly valuable for the early diagnosis of synucleinopathy [110], the importance of the resulting α-syn filaments is currently questionable. SAA was performed using sarkosyl-insoluble fractions extracted from three MSA cases used for cryo-EM study [48], and the structures of the SAA products were identified [111]. Some types of the amplified filaments resembled PF-IIB, but MSA filaments were not reproduced in SAA. Other types of filaments showed structures resembling those of spontaneously formed recombinant α-syn filaments. Cerebrospinal fluid collected from PD patients at various disease stages was also used for SAA, and the structures of the SAA products were reported [112]. As in the study described earlier, the majority of amplified α-syn filaments were identical to spontaneous recombinant α-syn filaments, and several other filaments identified at low percentages did not show the Lewy fold. The structures of α-syn filaments formed in other SAA studies also differed from those of brain-derived filaments [113-116]. More recently, it has been demonstrated that α-syn in serum from patients with synucleinopathy can be amplified by SAA after immunoprecipitation [117]. The ultrastructures of SAA products differed from those of brain-derived filaments, such as straight or twisted protofilaments in MSA-derived products and twisted or bundled filaments in LBD-derived products. Notably, the structures identified from SAA products do not necessarily reflect the structures of α-syn in body fluids. Since the α-syn concentration in body fluids is extremely low, at the picomolar level [118], the molecular species and structural details of body fluid-derived α-syn have not yet been fully established. Structural interpretation of α-syn filaments obtained from SAA using body fluids requires further characterization of the original seeds.
α-SYN FILAMENTS IN IN VITRO MODELS
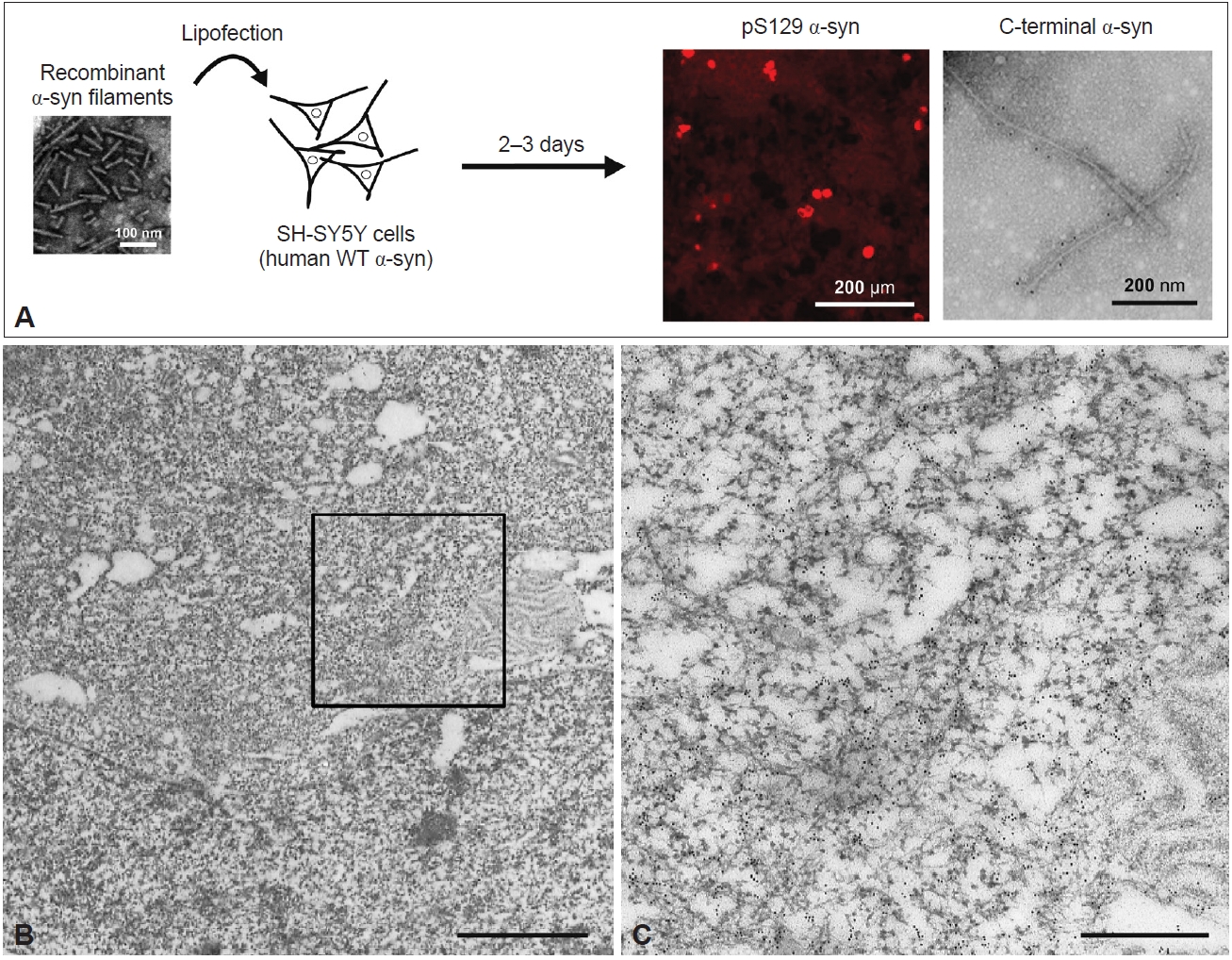
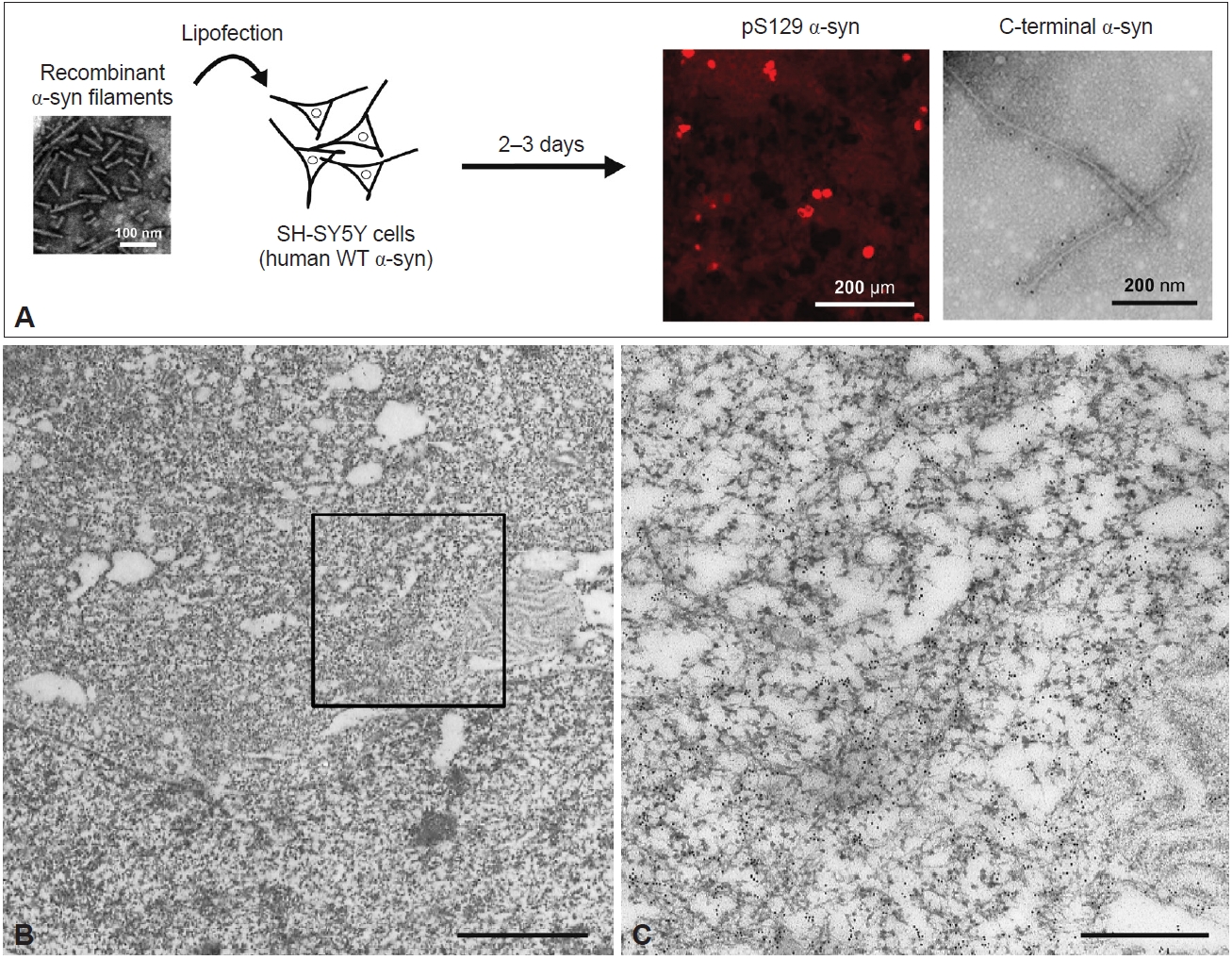
- As in in vitro studies, seeded α-syn aggregation also occurs in cultured cells and rodent primary cultures, and intracellular accumulation of filamentous α-syn is observed in seeded cells [119-122]. A few days after introduction of recombinant human α-syn filaments into neuroblastoma SH-SY5Y cells expressing human WT α-syn, introduced by lipofection, pS129-positive α-syn inclusions resembling those observed in the patient’s brain were formed (Figure 4A) [120]. These inclusions were also positive for ubiquitin, an autophagy substrate, p62, and thioflavin. Filamentous structures in the sarkosyl-insoluble fraction extracted from the seeded SH-SY5Y cells were labeled with pS129 and C-terminal α-syn antibodies (Figure 4A). Immuno-EM of the seeded cells showed that the α-syn inclusions consisted of filaments approximately 10 nm in diameter (Figure 4B, C). PS129-positive α-syn filaments were randomly oriented in the cytoplasm, formed reticular structures, and frequently were intermingled with mitochondria (Figure 4B, C). This presence of mitochondria in clustered α-syn filaments is a characteristic of LBs. Moreover, the introduction of recombinant α-syn filaments labeled with gold nanoparticles into SH-SY5Y cells resulted in polar filament elongation [123].
- Moreover, the addition of recombinant α-syn filaments to mouse primary neurons causes multiple types of insoluble α-syn pathology in a time-course-dependent manner [121]. On Day 7 after the seed treatment, small puncta and LN-like α-syn pathology with various lengths were observed, mainly in neurites, and immuno-EM revealed randomly organized or parallel-bundled pS129-positive α-syn filaments. Furthermore, larger α-syn inclusions were observed in the cytoplasm on Day 14, and some of these inclusions had an LB-like dense, round morphology. The seeded neurons at this time point showed abundant filamentous structures 14–16 nm in diameter in the cytoplasm, presynaptic terminals, postsynaptic terminals, and neurites. The interaction of α-syn filaments with organelles is suggested to occur along with the formation of cytoplasmic α-syn inclusions. Additional incubation led to the observation of LB-like α-syn inclusions distinguished by membranes on Day 21; these inclusions were composed not only of α-syn filaments but also of mitochondria, autophagosomes, and endolysosomal vesicles [124]. Immunocytochemistry showed that these LB-like α-syn inclusions were positive for an outer mitochondrial membrane protein, Tom 20, and a lysosome-associated membrane protein, LAMP1, and were partially colocalized with endoplasmic reticulum (ER) proteins. Cryo-electron tomography (ET) of the seeded rat primary neurons also revealed α-syn inclusions composed of abundant filaments, in which various organelles, such as ER, mitochondria, autophagolysosomal structures, and vesicles, were present [123]. Regarding cell-to-cell transmission, immuno-EM study of seeded human astrocytes showed that labeled recombinant α-syn filaments were observed in macropinosomes shortly after seed treatment, and some of them were transferred to neighboring naïve cells [125].
- Patient-derived α-syn filaments also cause the formation of pS129-positive filamentous inclusions in cultured cells and primary cultures. Remarkably, LBD-α-syn and MSA-α-syn exhibit distinct prion-like seeding properties. The seeding efficiency of LBD-α-syn is much less than that of recombinant α-syn filaments, while MSA-α-syn induces a greater extent of seeded aggregation, which is independent of the α-syn concentration in the seed fraction [71]. The unique seeding properties of patient-derived α-syn filaments are commonly observed in various types of cultured cells and in both primary neurons and oligodendrocytes, suggesting that distinct conformations of α-syn filaments differ in their ability to recruit normal α-syn [71,73,126,127]. Cryo-ET study showed that the addition of recombinant α-syn filaments or MSA-α-syn to rat primary neurons resulted in the formation of seeded α-syn filaments with different biophysical properties [123].
- These cellular studies demonstrated the formation of filamentous α-syn inclusions containing various organelles in the neuronal environment, akin to those of LBs observed in the patient’s brain. However, it remains unclear whether α-syn filaments formed in the cellular systems inherit the conformation of the original seeds. Cryo-EM structures of tau filaments extracted from SH-SY5Y cells seeded with patient-derived tau filaments appeared consistent with the possibility of templated tau filament amplification [128]. A similar approach is expected to elucidate the structure of cellular model-derived α-syn filaments and the contribution of the cellular environment to conformational templating.
α-SYN FILAMENTS IN CELLULAR MODELS
- Inoculation of recombinant and patient-derived α-syn filaments into the brains and peripheral tissues of rodents and nonhuman primates induces the formation of α-syn pathology followed by intracerebral spreading [129-137]. Although the distribution of spreading varies depending on the inoculation site [138-141], pS129- and thioflavin-positive neuronal α-syn pathology is observed in inoculated animal brains and peripheral tissues (Figure 5A). As in the cellular models, MSA-α-syn exhibits a greater ability to induce α-syn pathology than LBD-α-syn [71,73,142]. However, oligodendroglial α-syn pathology is not triggered even by the inoculation of MSA-α-syn. In the M83 transgenic (Tg) mouse line expressing human A53T α-syn, heterozygous mice inoculated with MSA-α-syn developed Gallyas-silver-negative neuronal α-syn pathology, which differs from the characteristics of GCIs [143,144]. This can be explained by the very low level of α-syn expression in oligodendrocytes. Inoculation of LBD-α-syn and MSA-α-syn into KOM2 Tg mice, which express α-syn in oligodendrocytes but not in neurons, resulted in the formation of oligodendroglial α-syn pathology [73]. This, together with the lack of α-syn pathology in seed-inoculated α-syn-knockout mice [130,131], supports the structural conversion of α-syn expressed in animals to a filament form. Recombinant α-syn filament strains have been reported to induce various types of neuronal α-syn pathology, heterogeneity of disease progression and cell death in rodent brains [90,145-147].
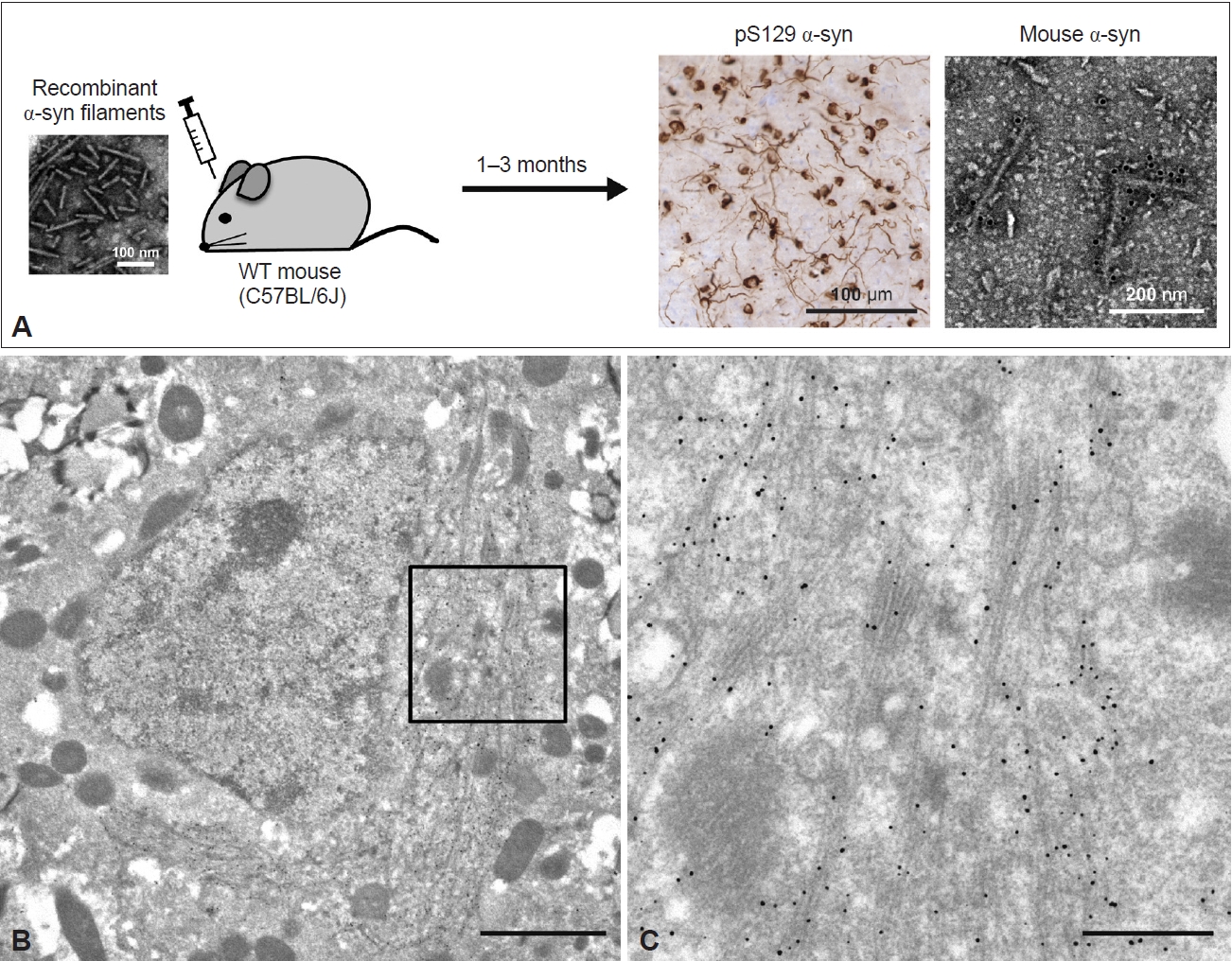
- Anti-α-syn-labeled filaments 10–16 nm in diameter were randomly present in the brain and spinal cord sections of M83 homozygous mice exhibiting α-syn pathology [148]. Detergent-insoluble fractions extracted from diseased M83 homozygous and seed-inoculated or seed-administered M83 heterozygous mice contained pS129-positive α-syn filaments [149,150]. Filamentous structures 5–10 nm in diameter were also observed in the sarkosyl-insoluble fraction extracted from WT mice inoculated with recombinant human α-syn filaments (Figure 5A) [151]. These filaments were recognized by mouse α-syn-specific and pS129 antibodies, indicating that recombinant human α-syn filaments recruited endogenous mouse α-syn into filaments. Immuno-EM of the inoculated WT mouse brain sections also showed the accumulation of numerous pS129-labeled elongated α-syn filaments 10–20 nm in diameter around the nucleus (Figure 5B, C). The α-syn filaments were present randomly or in parallel, partially as bundles, and intermingled with mitochondria and some types of vesicles (Figure 5B, C). This phenomenon resembles the development of α-syn pathology observed in LBD brains and in neuronal cellular models. While the cryo-EM structures of amyloid-β and tau filaments derived from Tg and knock-in mice have already been reported [152-154], the α-syn filaments derived from animal models have not yet been precisely characterized.
α-SYN FILAMENTS IN ANIMAL MODELS
- As we described above, the atomic structures of α-syn filaments extracted from synucleinopathy brains are not identical to those of α-syn filaments formed in vitro. What, other than mutations in α-syn, might cause these conformational differences? First, in vitro models do not fully mimic the cellular environment. The formation of distinct types of recombinant α-syn filaments under various conditions suggests that the initial misfolding of α-syn is easily influenced by external environmental factors [96,97,155]. Also, some additional densities identified in patient-derived α-syn and tau filaments have been speculated to be solvent molecules [58,156]. Since the initial misfolding form is supposed to be inherited in the subsequent templated amplification process, it is crucial to identify the cellular environmental factors and molecules involved in the formation of disease-associated α-syn folds. The presence of lipids in LBs suggests that some nonproteinaceous cofactors may affect not only filament formation but also the core structur [61,63]. LC‒MS/MS analysis revealed 296 proteins, including synaptic proteins, in LBs, indicating that protein-protein interactions would be important for LB formation [157]. Furthermore, the cell type-specific cellular environments of neurons and oligodendrocytes may contribute to the disease-specific α-syn folds. The failure to reproduce MSA filaments in vitro has suggested that oligodendroglial environment-derived factors may be essential not only in the initial misfolding but also in the templated amplification [111]. Diverse transcriptomic profiles have been reported between cell types and brain regions [158-160], suggesting the involvement of genetic factors and their changes with aging in cell type-specific environments. However, there is a critical question to be resolved in considering the importance of the oligodendroglial environment for the formation of the MSA folds. Although the α-syn expression level in oligodendrocytes has been reported to be increased in MSA brains [161], the actual expression level and the origin of α-syn constituting GCIs remain debatable [162]. If α-syn is transferred from neurons, what is the molecular species of the transferred α-syn? The obvious structural differences between LBD and MSA filaments militate against the possibility that filamentous α-syn is transferred from neurons to oligodendrocytes [48,58], instead favoring the transfer of monomeric or nonfilamentous oligomeric α-syn. Some types of cellular stress and neuronal impairment in the affected neurons may cause an increase in the SNCA mRNA level in oligodendrocytes and/or accelerate GCI formation. The experimental models mimicking neuronal α-syn pathology showed similar characteristics to LBs in LBD brains regarding the copresence of organelles, vesicles, and membrane structures with elongated α-syn filaments, which were also observed in neurons treated with MSA-α-syn [120,123,124,151]. These observations indicate that this is a hallmark of α-syn pathogenesis in neurons rather than a consequence of the type of α-syn seeds. Further studies are needed to elucidate in detail the α-syn pathogenesis in oligodendrocytes.
- In addition, the absence of post-translational modifications (PTMs) in in vitro models may influence the filament core structure. The additional densities in patient-derived α-syn filaments display similar biochemical characteristics [59], and PTMs might be involved in them. C-terminal phosphorylation modifies the negative charge of normal α-syn, increases the exposure of the N-terminal structure, and changes its interaction with environmental factors [163]. Phosphorylation at Y39 causes the formation of an electrostatic interaction network at the N-terminal region and changes the filament core structure, supporting the potential involvement of PTMs in α-syn filament formation [101]. Lysine PTMs are located on the outside of the structured filament core in synucleinopathy brains [48], suggesting that these modifications occur after the filament core is formed. However, the possible contributions of acetylation, ubiquitination and other PTMs need to be further investigated, and the PTM profiles of LBD and MSA should also be established in more detail. Immunoblot analysis of sarkoayl-insoluble α-syn derived from LBD and MSA brains show distinct high-molecular α-syn bands [71,164,165], indicating disease specificity in the PTM profile. In the case of tau, N-/C-terminal truncation has been shown to be essential for the in vitro formation of the folds seen in AD and chronic traumatic encephalopathy [156,166]. Disease-specific PK-resistant α-syn bands in LBD and MSA appear to be consistent with different C-terminal truncation processes [73,167].
IMPLICATIONS OF CONFORMATIONAL DIFFERENCES BETWEEN PATIENT- AND EXPERIMENTAL MODEL-DERIVED α-SYN FILAMENTS
- The ultrastructural and biochemical properties of pathological α-syn accumulated in synucleinopathy brains have been extensively characterized. Diseases in the same pathological category share structurally and biochemically identical α-syn filaments, implying that the conformation of α-syn filaments contributes to the pathological diversity of synucleinopathy. Cryo-EM studies of patient-derived α-syn filaments are consistent with prion-like, templated amplification of α-syn filaments in the human brain. However, the mechanisms by which α-syn adopts filamentstructural polymorphisms remain unclear. Conformational differences in α-syn filaments derived from patients’ brains and from experimental models indicate that environmental factors and molecules are involved in the formation of patient-derived α-syn folds, and these remain to be identified. More detailed biochemical characterization of LBD-α-syn and MSA-α-syn may be helpful in this regard. Recently, heterogeneity of in vitro and in vivo seeding properties triggered by α-syn seeds derived from patients with a single disease has been reported, suggesting a potential association with clinical phenotypes [113,168-171]. Although it is unknown whether there are structurally distinguishable subtypes within LBD or MSA, establishing links between seeding properties and other factors, including disease duration and progression, cognitive, motor and psychiatric symptoms and histopathological features, would further advance SAA-based early diagnosis. In addition, the patient-derived α-syn filament folds are expected to be helpful for positron emission tomography (PET) imaging and for developing therapeutic strategies. The atomic structure of recombinant α-syn filaments bound with a PET probe, [18F]-F0502B, has been reported [172], and the development of PET probes targeting the disease-specific filament core might enable discrimination between LBD and MSA at an early disease stage [173]. Similarly, the development of structure-based aggregation inhibitors and antibody therapies seems to be a promising avenue for future studies [174,175].
CONCLUSIONS
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This work was supported by Japan Science and Technology Agency, CREST Grant Number JPMJCR18H3 (to M.H.), Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP18ek0109391, JP-18dm0207019 (to M.H.), JSPS KAKENHI Grant Numbers JP20K16482 (to A.T.).
-
Author Contributions
Conceptualization: Airi Tarutani, Masato Hasegawa. Funding acquisition: Airi Tarutani, Masato Hasegawa. Investigation: Airi Tarutani, Masato Hasegawa. Writing—original draft: Airi Tarutani. Writing—review & editing: Masato Hasegawa.
Notes
- We are grateful to the patients and their families for making tissues available for research. Tissue samples used for immunohistochemistry and immuno-EM were supplied by the Japan Brain Banks at the Tokyo Metropolitan Institute of Geriatrics and Gerontology and Sagamihara National Hospital. Molecular graphics in Figure 3B were created with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Acknowledgments
- 1. Lewy FH. Paralysis agitans. I. Pathologische anatomie. In: Lewandowsky M, editor. Handbuch der Neurologie. Berlin: Spinger; 1912:920–933.
- 2. Tretiakoff C. Contribution à l'étude de l'anatomie pathologique du locus niger de Soemmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et de la maladie de Parkinson [dissertation]. Paris: University of Paris; 1919.
- 3. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872.PubMed
- 4. Kosaka K. Lewy bodies in cerebral cortex, report of three cases. Acta Neuropathol 1978;42:127–134.ArticlePubMedPDF
- 5. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–2047.ArticlePubMed
- 6. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature 1997;388:839–840.ArticlePubMedPDF
- 7. Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998;152:879–884.PubMedPMC
- 8. Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. α-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998;249:180–182.ArticlePubMed
- 9. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 10. Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet 1998;18:106–108.ArticlePubMedPDF
- 11. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–173.ArticlePubMed
- 12. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. α-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013;28:811–813.PubMed
- 13. Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013;73:459–471.ArticlePubMed
- 14. Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 2014;35:2180.e1–2180.e5.ArticlePubMed
- 15. Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, et al. Homozygous α-synuclein p.A53V in familial Parkinson’s disease. Neurobiol Aging 2017;57:248.e7–248.e12.PubMed
- 16. Kapasi A, Brosch JR, Nudelman KN, Agrawal S, Foroud TM, Schneider JA. A novel SNCA E83Q mutation in a case of dementia with Lewy bodies and atypical frontotemporal lobar degeneration. Neuropathology 2020;40:620–626.ArticlePubMedPMCPDF
- 17. Liu H, Koros C, Strohäker T, Schulte C, Bozi M, Varvaresos S, et al. A novel SNCA A30G mutation causes familial Parkinson’s disease. Mov Disord 2021;36:1624–1633.ArticlePubMedPDF
- 18. Fevga C, Park Y, Lohmann E, Kievit AJ, Breedveld GJ, Ferraro F, et al. A new α-synuclein missense variant (Thr72Met) in two Turkish families with Parkinson’s disease. Parkinsonism Relat Disord 2021;89:63–72.PubMedPMC
- 19. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841.ArticlePubMed
- 20. Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 2016;22(Suppl 1):S1–S6.ArticlePubMed
- 21. Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett 1994;345:27–32.ArticlePubMedPDF
- 22. Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998;273:9443–9449.ArticlePubMed
- 23. Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:11282–11286.ArticlePubMedPMC
- 24. Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem 2001;276:2380–2386.ArticlePubMed
- 25. Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM. Enhanced vulnerability to oxidative stress by α-synuclein mutations and C-terminal truncation. Neuroscience 2000;97:279–284.ArticlePubMed
- 26. Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, et al. NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J 2004;23:2039–2046.ArticlePubMedPMC
- 27. Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, et al. Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s diseaselinked mutations. Proc Natl Acad Sci U S A 2005;102:2162–2167.ArticlePubMedPMC
- 28. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988;8:2804–2815.ArticlePubMedPMC
- 29. Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239–252.ArticlePubMed
- 30. Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, et al. Double-knockout mice for α- and β-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A 2004;101:14966–14971.ArticlePubMedPMC
- 31. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010;329:1663–1667.ArticlePubMedPMC
- 32. Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005;123:383–396.ArticlePubMed
- 33. Castagnet PI, Golovko MY, Barceló-Coblijn GC, Nussbaum RL, Murphy EJ. Fatty acid incorporation is decreased in astrocytes cultured from α-synuclein gene-ablated mice. J Neurochem 2005;94:839–849.ArticlePubMed
- 34. Rappley I, Myers DS, Milne SB, Ivanova PT, Lavoie MJ, Brown HA, et al. Lipidomic profiling in mouse brain reveals differences between ages and genders, with smaller changes associated with α-synuclein genotype. J Neurochem 2009;111:15–25.ArticlePubMedPMC
- 35. Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, et al. Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol Cell 2019;73:1001–1014.E8.ArticlePubMed
- 36. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361:1651–1661.ArticlePubMedPMC
- 37. Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011;89:168–175.ArticlePubMedPMC
- 38. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996;35:13709–13715.ArticlePubMed
- 39. Iwatsubo T, Yamaguchi H, Fujimuro M, Yokosawa H, Ihara Y, Trojanowski JQ, et al. Purification and characterization of Lewy bodies from the brains of patients with diffuse Lewy body disease. Am J Pathol 1996;148:1517-1529. Erratum in: Am J Pathol 1996;149:1770-1771. Erratum in: Am J Pathol 1997;150:2255.
- 40. Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jäkälä P, et al. The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J Neurochem 2001;76:87–96.ArticlePubMed
- 41. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like crossbeta conformation. Proc Natl Acad Sci U S A 2000;97:4897–4902.PubMedPMC
- 42. Miake H, Mizusawa H, Iwatsubo T, Hasegawa M. Biochemical characterization of the core structure of α-synuclein filaments. J Biol Chem 2002;277:19213–19219.ArticlePubMed
- 43. Cantuti-Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, et al. α-synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol 2005;64:1058–1066.PubMed
- 44. Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998;95:13363–13383.ArticlePubMedPMC
- 45. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002;4:160–164.ArticlePubMedPDF
- 46. Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, et al. Phosphorylated α-synuclein is ubiquitinated in αsynucleinopathy lesions. J Biol Chem 2002;277:49071–49076.ArticlePubMed
- 47. Schmid AW, Fauvet B, Moniatte M, Lashuel HA. α-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol Cell Proteomics 2013;12:3543–3558.PubMedPMC
- 48. Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020;585:464–469.ArticlePubMedPMCPDF
- 49. Zhang S, Zhu R, Pan B, Xu H, Olufemi MF, Gathagan RJ, et al. Posttranslational modifications of soluble α-synuclein regulate the amplification of pathological α-synuclein. Nat Neurosci 2023;26:213–225.ArticlePubMedPMCPDF
- 50. Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013;501:45–51.ArticlePubMedPMCPDF
- 51. Goedert M. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015;349:1255555.ArticlePubMed
- 52. Tarutani A, Hasegawa M. Prion-like propagation of α-synuclein in neurodegenerative diseases. Prog Mol Biol Transl Sci 2019;168:323–348.ArticlePubMed
- 53. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211.ArticlePubMed
- 54. Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, et al. Accumulation of phosphorylated α-synuclein in aging human brain. J Neuropathol Exp Neurol 2003;62:644–654.ArticlePubMed
- 55. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008;14:501–503.ArticlePubMedPDF
- 56. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 2008;14:504–506.ArticlePubMedPDF
- 57. Li W, Englund E, Widner H, Mattsson B, van Westen D, Lätt J, et al. Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A 2016;113:6544–6549.ArticlePubMedPMC
- 58. Yang Y, Shi Y, Schweighauser M, Zhang X, Kotecha A, Murzin AG, et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 2022;610:791–795.ArticlePubMedPDF
- 59. Yang Y, Garringer HJ, Shi Y, Lövestam S, Peak-Chew S, Zhang X, et al. New SNCA mutation and structures of α-synuclein filaments from juvenile-onset synucleinopathy. Acta Neuropathol 2023;145:561–572.ArticlePubMedPMCPDF
- 60. Duffy PE, Tennyson VM. Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus caeruleus in Parkinson’s disease. J Neuropathol Exp Neurol 1965;24:398–414.Article
- 61. Forno LS. Concentric hyalin intraneuronal inclusions of Lewy type in the brains of elderly persons (50 incidental cases): relationship to parkinsonism. J Am Geriatr Soc 1969;17:557–575.ArticlePubMed
- 62. Forno LS, Norville RL. Ultrastructure of Lewy bodies in the stellate ganglion. Acta Neuropathol 1976;34:183–197.ArticlePubMedPDF
- 63. Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 2019;22:1099–1109.ArticlePubMedPDF
- 64. Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol 1988;75:345–353.ArticlePubMedPDF
- 65. Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol 2013;47:495–508.ArticlePubMedPDF
- 66. Fares MB, Jagannath S, Lashuel HA. Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 2021;22:111–131.ArticlePubMedPDF
- 67. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989;94(1-3):79–100.ArticlePubMed
- 68. Arima K, Murayama S, Mukoyama M, Inose T. Immunocytochemical and ultrastructural studies of neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 1. Neuronal cytoplasmic inclusions. Acta Neuropathol 1992;83:453–460.ArticlePubMedPDF
- 69. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 1998;95:6469–6473.ArticlePubMedPMC
- 70. Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205–208.ArticlePubMed
- 71. Tarutani A, Arai T, Murayama S, Hisanaga SI, Hasegawa M. Potent prion-like behaviors of pathogenic α-synuclein and evaluation of inactivation methods. Acta Neuropathol Commun 2018;6:29.ArticlePubMedPMCPDF
- 72. Crowther RA, Daniel SE, Goedert M. Characterisation of isolated α-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci Lett 2000;292:128–130.ArticlePubMed
- 73. Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018;557:558–563.ArticlePubMedPMCPDF
- 74. Moors TE, Mona D, Luehe S, Duran-Pacheco G, Spycher L, Mundigl O, et al. Multi-platform quantitation of α-synuclein human brain proteoforms suggests disease-specific biochemical profiles of synucleinopathies. Acta Neuropathol Commun 2022;10:82.PubMedPMC
- 75. Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 1984;87:848–856.ArticlePubMed
- 76. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 2010;119:689–702.ArticlePubMedPMCPDF
- 77. Sanchez-Ferro Á, Rabano A, Catalán MJ, Rodríguez-Valcárcel FC, Fernández Díez S, Herreros-Rodríguez J, et al. In vivo gastric detection of α-synuclein inclusions in Parkinson’s disease. Mov Disord 2015;30:517–524.ArticlePubMed
- 78. Tanei ZI, Saito Y, Ito S, Matsubara T, Motoda A, Yamazaki M, et al. Lewy pathology of the esophagus correlates with the progression of Lewy body disease: a Japanese cohort study of autopsy cases. Acta Neuropathol 2021;141:25–37.ArticlePubMedPDF
- 79. Takao M, Ghetti B, Yoshida H, Piccardo P, Narain Y, Murrell JR, et al. Early-onset dementia with Lewy bodies. Brain Pathol 2004;14:137–147.ArticlePubMed
- 80. Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem 1999;274:7619–7622.ArticlePubMed
- 81. Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, et al. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett 2004;576:363–368.ArticlePubMedPDF
- 82. Yonetani M, Nonaka T, Masuda M, Inukai Y, Oikawa T, Hisanaga S, et al. Conversion of wild-type α-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem 2009;284:7940–7950.ArticlePubMedPMC
- 83. Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovičić A, Kiely A, et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet 2014;23:4491–4509.ArticlePubMedPMC
- 84. Mohite GM, Kumar R, Panigrahi R, Navalkar A, Singh N, Datta D, et al. Comparison of kinetics, toxicity, oligomer formation, and membrane binding capacity of α-synuclein familial mutations at the A53 site, including the newly discovered A53V mutation. Biochemistry 2018;57:5183–5187.ArticlePubMed
- 85. Kumar ST, Mahul-Mellier AL, Hegde RN, Rivière G, Moons R, Ibáñez de Opakua A, et al. A NAC domain mutation (E83Q) unlocks the pathogenicity of human α-synuclein and recapitulates its pathological diversity. Sci Adv 2022;8:eabn0044.PubMedPMC
- 86. Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett 1998;436:309–312.ArticlePubMedPDF
- 87. Terada M, Suzuki G, Nonaka T, Kametani F, Tamaoka A, Hasegawa M. The effect of truncation on prion-like properties of α-synuclein. J Biol Chem 2018;293:13910–13920.ArticlePubMedPMC
- 88. Zhang C, Pei Y, Zhang Z, Xu L, Liu X, Jiang L, et al. C-terminal truncation modulates α-synuclein’s cytotoxicity and aggregation by promoting the interactions with membrane and chaperone. Commun Biol 2022;5:798.ArticlePubMedPMCPDF
- 89. Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two α-synuclein strains. Nat Commun 2013;4:2575.PubMed
- 90. Suzuki G, Imura S, Hosokawa M, Katsumata R, Nonaka T, Hisanaga SI, et al. α-synuclein strains that cause distinct pathologies differentially inhibit proteasome. Elife 2020;9:e56825. ArticlePubMedPMCPDF
- 91. Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from α-synuclein in vitro. Biochemistry 2002;41:1502–1511.ArticlePubMed
- 92. Byrd EJ, Wilkinson M, Radford SE, Sobott F. Taking Charge: Metal Ions Accelerate Amyloid Aggregation in Sequence Variants of α-synuclein. J Am Soc Mass Spectrom 2023;34:493–504.ArticlePubMedPMCPDF
- 93. Dasari AKR, Dillard L, Yi S, Viverette E, Hojjatian A, Sengupta U, et al. Untwisted α-synuclein Filaments Formed in the Presence of Lipid Vesicles. Biochemistry 2022;61:1766–1773.ArticlePubMedPDF
- 94. Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, et al. Cryo-EM structure of α-synuclein fibrils. Elife 2018;7:e36402. PubMedPMC
- 95. Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, et al. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res 2018;28:897–903.ArticlePubMedPMCPDF
- 96. Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 2018;9:3609.ArticlePubMedPMCPDF
- 97. Guerrero-Ferreira R, Taylor NM, Arteni AA, Kumari P, Mona D, Ringler P, et al. Two new polymorphic structures of human full-length α-synuclein fibrils solved by cryo-electron microscopy. Elife 2019;8:e48907. PubMedPMC
- 98. Ni X, McGlinchey RP, Jiang J, Lee JC. Structural insights into α-synuclein fibril polymorphism: effects of Parkinson’s disease-related C-terminal truncations. J Mol Biol 2019;431:3913–3919.ArticlePubMedPMC
- 99. Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, et al. Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat Struct Mol Biol 2019;26:1044–1052.ArticlePubMedPMCPDF
- 100. Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat Commun 2020;11:2643.ArticlePubMedPMCPDF
- 101. Zhao K, Lim YJ, Liu Z, Long H, Sun Y, Hu JJ, et al. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc Natl Acad Sci U S A 2020;117:20305–20315.ArticlePubMedPMC
- 102. Boyer DR, Li B, Sun C, Fan W, Zhou K, Hughes MP, et al. The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc Natl Acad Sci U S A 2020;117:3592–3602.ArticlePubMedPMC
- 103. Sun Y, Hou S, Zhao K, Long H, Liu Z, Gao J, et al. Cryo-EM structure of full-length α-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res 2020;30:360–362.ArticlePubMedPMCPDF
- 104. Sun Y, Long H, Xia W, Wang K, Zhang X, Sun B, et al. The hereditary mutation G51D unlocks a distinct fibril strain transmissible to wildtype α-synuclein. Nat Commun 2021;12:6252.ArticlePubMedPMCPDF
- 105. Sun C, Zhou K, DePaola P IV, Shin WS, Hillyer T, Sawaya MR, et al. Cryo-EM structure of amyloid fibril formed by α-synuclein hereditary A53E mutation reveals a distinct protofilament interface. J Biol Chem 2023;299:104566.ArticlePubMedPMC
- 106. Concha-Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed amplification assay for the detection of pathologic α-synuclein aggregates in cerebrospinal fluid. Nat Protoc 2023;18:1179–1196.ArticlePubMedPMCPDF
- 107. Kuzkina A, Rößle J, Seger A, Panzer C, Kohl A, Maltese V, et al. Combining skin and olfactory α-synuclein seed amplification assays (SAA)- towards biomarker-driven phenotyping in synucleinopathies. NPJ Parkinsons Dis 2023;9:79.ArticlePubMedPMCPDF
- 108. Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020;578:273–277.ArticlePubMedPMCPDF
- 109. Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orrù CD, et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 2020;140:49–62.ArticlePubMedPMCPDF
- 110. Siderowf A, Concha-Marambio L, Lafontant DE, Farris CM, Ma Y, Urenia PA, et al. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol 2023;22:407–417.ArticlePubMedPMC
- 111. Lovestam S, Schweighauser M, Matsubara T, Murayama S, Tomita T, Ando T, et al. Seeded assembly in vitro does not replicate the structures of α-synuclein filaments from multiple system atrophy. FEBS Open Bio 2021;11:999–1013.ArticlePubMedPMCPDF
- 112. Fan Y, Sun Y, Yu W, Tao Y, Xia W, Liu Y, et al. Conformational change of α-synuclein fibrils in cerebrospinal fluid from different clinical phases of Parkinson’s disease. Structure 2023;31:78–87.e5.ArticlePubMed
- 113. Strohäker T, Jung BC, Liou SH, Fernandez CO, Riedel D, Becker S, et al. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat Commun 2019;10:5535.ArticlePubMedPMCPDF
- 114. Burger D, Fenyi A, Bousset L, Stahlberg H, Melki R. Cryo-EM structure of α-synuclein fibrils amplified by PMCA from PD and MSA patient brains. BioRxiv 451588 [Preprint] 2021;[cited 2021 Jul 9]. Available at: https://doi.org/10.1101/2021.07.08.451588. Article
- 115. Dhavale DD, Barclay AM, Borcik CG, Basore K, Gordon IR, Liu J, et al. Structure of α-synuclein fibrils derived from human Lewy body dementia tissue. BioRxiv 523303 [Preprint] 2023;[cited 2023 Jan 10]. Available at: https://doi.org/10.1101/2023.01.09.523303. Article
- 116. Sokratian A, Zhou Y, Xu E, Viverette E, Dillard L, Yuan Y, et al. Structural and functional landscape of α-synuclein fibril conformations amplified from cerebrospinal fluid. BioRxiv 499896 [Preprint] 2022;[cited 2022 Jul 13]. Available at: https://doi.org/10.1101/2022.07.13.499896. Article
- 117. Okuzumi A, Hatano T, Matsumoto G, Nojiri S, Ueno SI, ImamichiTatano Y, et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med 2023;29:1448–1455.ArticlePubMedPMCPDF
- 118. Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T, et al. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 2010;75:1766–1772.ArticlePubMed
- 119. Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009;106:20051–20056.ArticlePubMedPMC
- 120. Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 2010;285:34885–34898.PubMedPMC
- 121. Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011;72:57–71.ArticlePubMedPMC
- 122. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci U S A 2009;106:13010–13015.ArticlePubMedPMC
- 123. Trinkaus VA, Riera-Tur I, Martinez-Sánchez A, Bauerlein FJB, Guo Q, Arzberger T, et al. In situ architecture of neuronal α-synuclein inclusions. Nat Commun 2021;12:2110.ArticlePubMedPMCPDF
- 124. Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, et al. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 2020;117:4971–4982.ArticlePubMedPMC
- 125. Bayati A, Banks E, Han C, Luo W, Reintsch WE, Zorca CE, et al. Rapid macropinocytic transfer of α-synuclein to lysosomes. Cell Rep 2022;40:111102.ArticlePubMed
- 126. Woerman AL, Stöhr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A 2015;112:E4949–E4958.ArticlePubMedPMC
- 127. Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, et al. Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics. J Biol Chem 2019;294:1045–1058.ArticlePubMed
- 128. Tarutani A, Lövestam S, Zhang X, Kotecha A, Robinson AC, Mann DMA, et al. Cryo-EM structures of tau filaments from SH-SY5Y cells seeded with brain extracts from cases of Alzheimer’s disease and corticobasal degeneration. FEBS Open Bio 2023;13:1394–1404.ArticlePubMedPMC
- 129. Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 2012;209:975–986.ArticlePubMedPMCPDF
- 130. Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949–953.ArticlePubMedPMC
- 131. Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological α-synuclein in brain. Brain 2013;136:1128–1138.ArticlePubMedPMC
- 132. Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 2013;110:19555–19560.ArticlePubMedPMC
- 133. Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014;75:351–362.ArticlePubMed
- 134. Bernis ME, Babila JT, Breid S, Wusten KA, Wüllner U, Tamgüney G. Prion-like propagation of human brain-derived α-synuclein in transgenic mice expressing human wild-type α-synuclein. Acta Neuropathol Commun 2015;3:75.PubMedPMC
- 135. Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 2015;82:185–199.ArticlePubMedPMC
- 136. Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, et al. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol Commun 2017;5:12.ArticlePubMedPMCPDF
- 137. Kawakami I, Motoda A, Hashimoto M, Shimozawa A, Masuda-Suzukake M, Ohtani R, et al. Progression of phosphorylated α-synuclein in Macaca fuscata. Brain Pathol 2021;31:e12952.ArticlePubMedPMCPDF
- 138. Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 2013;126:555–573.ArticlePubMedPMCPDF
- 139. Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, et al. Pathological α-synuclein propagates through neural networks. Acta Neuropathol Commun 2014;2:88.PubMedPMC
- 140. Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 2018;13:21.ArticlePubMedPMCPDF
- 141. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 2020;23:327–336.ArticlePubMedPMCPDF
- 142. Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 2015;112:E5308–E5317.ArticlePubMedPMC
- 143. Lavenir I, Passarella D, Masuda-Suzukake M, Curry A, Holton JL, Ghetti B, et al. Silver staining (Campbell-Switzer) of neuronal α-synuclein assemblies induced by multiple system atrophy and Parkinson’s disease brain extracts in transgenic mice. Acta Neuropathol Commun 2019;7:148.ArticlePubMedPMCPDF
- 144. Uchihara T, Nakamura A, Mochizuki Y, Hayashi M, Orimo S, Isozaki E, et al. Silver stainings distinguish Lewy bodies and glial cytoplasmic inclusions: comparison between Gallyas-Braak and Campbell-Switzer methods. Acta Neuropathol 2005;110:255–260.ArticlePubMedPDF
- 145. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015;522:340–344.ArticlePubMedPDF
- 146. Rey NL, Bousset L, George S, Madaj Z, Meyerdirk L, Schulz E, et al. α-synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun 2019;7:221.ArticlePubMedPMCPDF
- 147. Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC, et al. α-synuclein strains target distinct brain regions and cell types. Nat Neurosci 2020;23:21–31.ArticlePubMedPDF
- 148. Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002;34:521–533.ArticlePubMed
- 149. Morgan SA, Lavenir I, Fan J, Masuda-Suzukake M, Passarella D, DeTure MA, et al. α-synuclein filaments from transgenic mouse and human synucleinopathy-containing brains are major seed-competent species. J Biol Chem 2020;295:6652–6664.ArticlePubMedPMC
- 150. Macdonald JA, Chen JL, Masuda-Suzukake M, Schweighauser M, Jaunmuktane Z, Warner T, et al. Assembly of α-synuclein and neurodegeneration in the central nervous system of heterozygous M83 mice following the peripheral administration of α-synuclein seeds. Acta Neuropathol Commun 2021;9:189.ArticlePubMedPMCPDF
- 151. Tarutani A, Suzuki G, Shimozawa A, Nonaka T, Akiyama H, Hisanaga S, et al. The effect of fragmented pathogenic α-synuclein seeds on prionlike propagation. J Biol Chem 2016;291:18675–18688.ArticlePubMedPMC
- 152. Yang Y, Arseni D, Zhang W, Huang M, Lövestam S, Schweighauser M, et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022;375:167–172.ArticlePubMedPMC
- 153. Yang Y, Zhang W, Murzin AG, Schweighauser M, Huang M, Lövestam S, et al. Cryo-EM structures of amyloid-β filaments with the Arctic mutation (E22G) from human and mouse brains. Acta Neuropathol 2023;145:325–333.ArticlePubMedPMCPDF
- 154. Schweighauser M, Murzin AG, Macdonald J, Lavenir I, Crowther RA, Scheres SHW, et al. Cryo-EM structures of tau filaments from the brains of mice transgenic for human mutant P301S Tau. Acta Neuropathol Commun 2023;11:160.ArticlePubMedPMCPDF
- 155. Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol 2016;23:409–415.ArticlePubMedPMCPDF
- 156. Lövestam S, Koh FA, van Knippenberg B, Kotecha A, Murzin AG, Goedert M, et al. Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife 2022;11:e76494. PubMedPMC
- 157. Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, et al. Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathol 2007;17:139–145.ArticlePubMedPMC
- 158. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 2008;28:264–278.ArticlePubMedPMC
- 159. Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 2015;112:7285–7290.ArticlePubMedPMC
- 160. Sadeghi I, Gispert JD, Palumbo E, Muñoz-Aguirre M, Wucher V, D’Argenio V, et al. Brain transcriptomic profiling reveals common alterations across neurodegenerative and psychiatric disorders. Comput Struct Biotechnol J 2022;20:4549–4561.ArticlePubMedPMC
- 161. Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, et al. α-synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014;62:964–970.ArticlePubMedPMCPDF
- 162. Valera E, Masliah E. The neuropathology of multiple system atrophy and its therapeutic implications. Auton Neurosci 2018;211:1–6.ArticlePubMed
- 163. Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, et al. Extent of N-terminus exposure of monomeric α-synuclein determines its aggregation propensity. Nat Commun 2020;11:2820.PubMedPMC
- 164. Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem 2006;281:29739–29752.ArticlePubMed
- 165. Tarutani A, Adachi T, Akatsu H, Hashizume Y, Hasegawa K, Saito Y, et al. Ultrastructural and biochemical classification of pathogenic tau, α-synuclein and TDP-43. Acta Neuropathol 2022;143:613–640.ArticlePubMedPMCPDF
- 166. Al-Hilaly YK, Pollack SJ, Vadukul DM, Citossi F, Rickard JE, Simpson M, et al. Alzheimer’s disease-like paired helical filament assembly from truncated tau protein is independent of disulfide crosslinking. J Mol Biol 2017;429:3650–3665.ArticlePubMed
- 167. Hass EW, Sorrentino ZA, Xia Y, Lloyd GM, Trojanowski JQ, Prokop S, et al. Disease-, region- and cell type specific diversity of α-synuclein carboxy terminal truncations in synucleinopathies. Acta Neuropathol Commun 2021;9:146.ArticlePubMedPMCPDF
- 168. Sokratian A, Ziaee J, Kelly K, Chang A, Bryant N, Wang S, et al. Heterogeneity in α-synuclein fibril activity correlates to disease phenotypes in Lewy body dementia. Acta Neuropathol 2021;141:547–564.ArticlePubMedPMCPDF
- 169. Martinez-Valbuena I, Swinkin E, Santamaria E, Fernandez-Irigoyen J, Sackmann V, Kim A, et al. α-synuclein molecular behavior and nigral proteomic profiling distinguish subtypes of Lewy body disorders. Acta Neuropathol 2022;144:167–185.ArticlePubMedPDF
- 170. Martinez-Valbuena I, Visanji NP, Kim A, Lau HHC, So RWL, Alshimemeri S, et al. α-synuclein seeding shows a wide heterogeneity in multiple system atrophy. Transl Neurodegener 2022;11:7.PubMedPMC
- 171. Zhang S, Dauer K, Strohäker T, Tatenhorst L, Caldi Gomes L, Mayer S, et al. α-synuclein fibrils amplified from multiple system atrophy and Parkinson’s disease patient brain spread after intracerebral injection into mouse brain. Brain Pathol 2023;33:e13196. PubMedPMC
- 172. Xiang J, Tao Y, Xia Y, Luo S, Zhao Q, Li B, et al. Development of an α-synuclein positron emission tomography tracer for imaging synucleinopathies. Cell 2023;186:3350–3367.e19.ArticlePubMed
- 173. Smith R, Capotosti F, Schain M, Ohlsson T, Vokali E, Molette J, et al. The α-synuclein PET tracer [18F] ACI-12589 distinguishes multiple system atrophy from other neurodegenerative diseases. Nat Commun 2023;14:6750.ArticlePubMedPMCPDF
- 174. Murray KA, Hu CJ, Pan H, Lu J, Abskharon R, Bowler JT, et al. Small molecules disaggregate α-synuclein and prevent seeding from patient brain-derived fibrils. Proc Natl Acad Sci U S A 2023;120:e2217835120. PubMedPMC
- 175. Chia S, Faidon Brotzakis Z, Horne RI, Possenti A, Mannini B, Cataldi R, et al. Structure-based discovery of small-molecule inhibitors of the autocatalytic proliferation of α-synuclein aggregates. Mol Pharm 2023;20:183–193.ArticlePubMedPDF
- 176. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci 2023;20:e4792.
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite