E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 17(1); 2024 > Article
-
Case Report
A New Phenotype of TUBB4A Mutation in a Family With Adult-Onset Progressive Spastic Paraplegia and Isolated Hypomyelination Leukodystrophy: A Case Report and Literature Review -
Pei-Chen Hsieh1
 , Pei Shan Yu1, Wen-Lang Fan2,3, Chun-Chieh Wang1, Chih-Ying Chao1, Yih-Ru Wu1,4
, Pei Shan Yu1, Wen-Lang Fan2,3, Chun-Chieh Wang1, Chih-Ying Chao1, Yih-Ru Wu1,4
-
Journal of Movement Disorders 2024;17(1):94-98.
DOI: https://doi.org/10.14802/jmd.23142
Published online: October 23, 2023
1Department of Neurology, Chang Gung Memorial Hospital, Linkou Medical Center, Taoyuan, Taiwan
2Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
3Genomic Medicine Core Laboratory, Chang Gung Memorial Hospital, Linkou Medical Center, Taoyuan, Taiwan
4Department of Neurology, College of Medicine, Chang Gung University, Taoyuan, Taiwan
- Corresponding author: Yih-Ru Wu, MD Department of Neurology, Chang Gung Memorial Hospital, Linkou Medical Center, College of Medicine, Chang Gung University, No. 5, Fuxing Street, Taoyuan 33305, Taiwan / Tel: +886-3-3281200 Ext. 8349 / Fax: +886-3-328722 / E-mail: yihruwu@cgmh.org.tw
Copyright © 2024 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 816 Views
- 106 Download
ABSTRACT
- Tubulin beta 4A class IVa (TUBB4A) spectrum disorders include autosomal dominant dystonia type 4 or hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC syndrome). However, in rare cases, only mild hypomyelination in the cortex with no basal ganglia atrophy may be observed. We report a case of a family with TUBB4A mutation and complicated hereditary spasticity paraplegia (HSP). We performed quadro whole-exome sequencing (WES) on the family to identify the causative gene of progressive spastic paraparesis with isolated hypomyelination leukodystrophy. We identified a novel TUBB4A p.F341L mutation, which was present in all three affected patients but absent in the unaffected father. The affected patients presented with adult-onset TUBB4A disorder, predominant spastic paraparesis with/without ataxia, and brain hypomyelination with no cognitive impairment or extrapyramidal symptoms. In the literature, HSP is considered a TUBB4A spectrum disorder.
- TUBB4A gene–related disorders have a broad clinical spectrum, with manifestations including dystonia, spastic paraplegia, cognitive decline, and cerebellar ataxia [1,2]. TUBB4A encodes a member of the β-tubulin protein family primarily expressed in the cerebellar, basal ganglia, and white matter regions [1]. Common findings in brain magnetic resonance imaging (MRI) include diffuse hyperintensity in white matter on T2-weighted images and mild hypo- or isointensity relative to gray matter structures on T1-weighted images. Additionally, progressive atrophy in the basal ganglia and cerebellum may be evident. This condition is referred to as hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC syndrome). According to Kancheva et al., TUBB4A mutation may present as one or more spectrum disorder(s) without basal ganglia atrophy or obvious cognitive impairment [3,4]. Herein, we report a novel missense mutation of TUBB4A in a family that presented with complicated hereditary spasticity paraplegia (HSP).
INTRODUCTION
- Clinical report
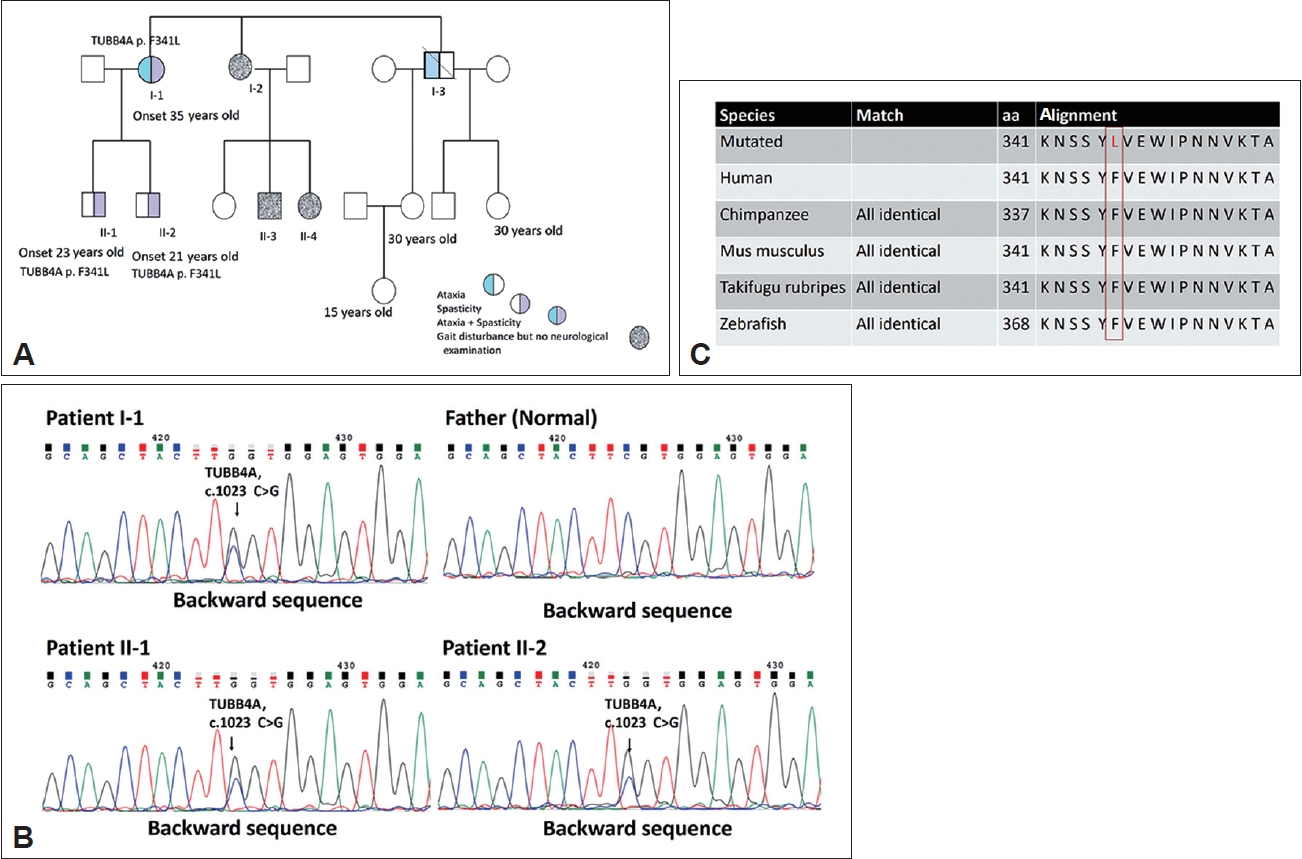
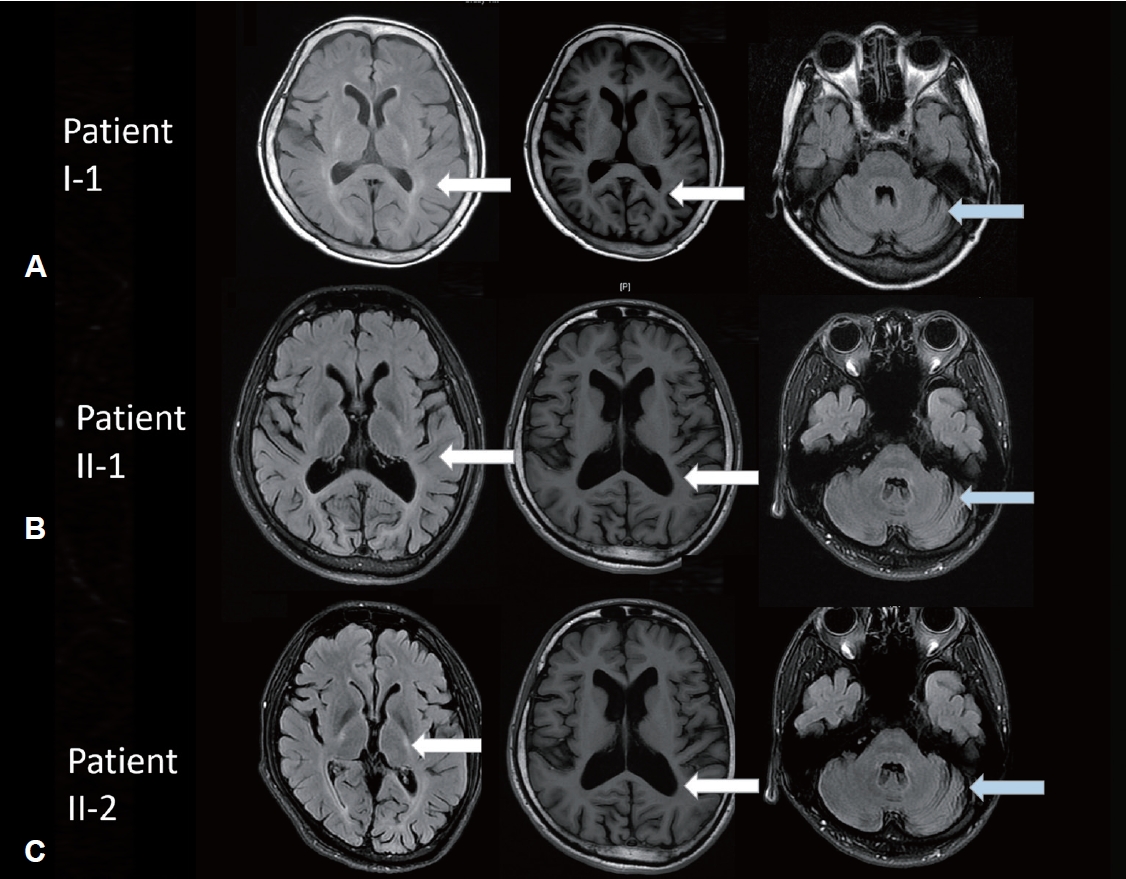
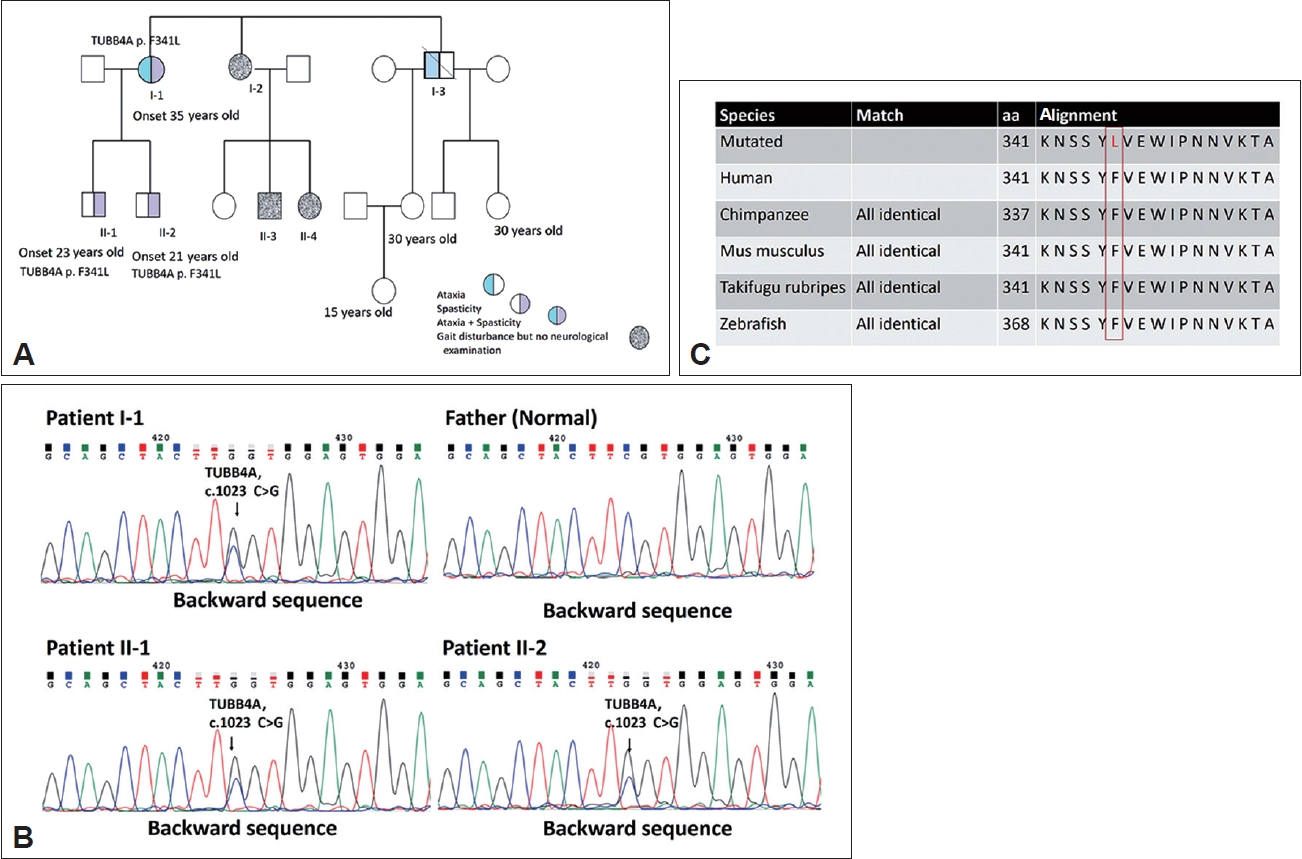
- The proband, a 56-year-old woman, experienced the onset of ataxia at the age of 35, which was subsequently accompanied by severe spasticity in both lower limbs. Her symptoms gradually worsened over time. A neurological examination revealed decreased muscle strength in the distal muscles of the lower extremities and normal finger-nose-finger test, saccade, and pursuit but impaired joint position and vibration sense. The gait pattern was a wide base with spasticity. The levels of deep tendon reflexes in all four limbs were increased, and the Babinski sign was positive 10 years after symptom onset. The proband had a score of 28 of 30 on the Mini-Mental State Examination (MMSE) and a score of 91 of 100 on the Cognitive Ability Screening Instrument (CASI). The motor-evoked potential (MEP) analysis indicated a lack of response following bilateral hemisphere stimulation, potentially indicating poor bilateral motor cortex excitability in the leg area. The visual-evoked potential (VEP) analysis indicated a prolonged peak latency of P100 in both eyes. Her brain MRI indicated predominant cerebellar atrophy and hyperintensity in the T2 image over the bilateral posterior internal capsule and periventricular area, with isointensity in the T1 image. In addition, her two children, sister and brother, had progressive gait disturbance (Figure 1A). However, her two children (II-1 and II-2) exhibited normal motor milestones in early childhood.
- Patients II-1 and II-2 had progressive onset of spastic gait and experienced frequent falls beginning at the ages of 23 and 21, respectively. Neurological examinations were performed at ages 35 and 31, respectively, which revealed mild dysarthria, normal muscle strength, increased deep tendon reflex, and the presence of the Babinski sign on both sides of the body. All sensory tests, such as the pinprick, joint position, and vibration sense assessments, yielded normal results. The cerebellar test showed normal saccade, pursuit, and eye movement and no signs of ataxia or dysmetria. Dystonia or other parkinsonism was not observed. Patients II-1 and II-2 obtained identical scores on the MMSE, achieving a score of 28 out of 30. Furthermore, their CASI scores were 98 out of 100 for Patient II-1 and 95 out of 100 for Patient II-2. Both siblings had prolonged VEPs in both eyes and poor excitability of the bilateral motor cortex in the MEP test. The nerve conduction study revealed bilateral mild median nerve neuropathy across the wrist in Patient II-1 and normal results in Patient II-2. Brain MRIs were also performed, which demonstrated 1) T2 hyperintensity and T1 isointensity in the posterior internal capsule and periventricular area and 2) cortical atrophy (Figure 2).
- Pathogenic variant and family cosegregation analysis
- To detect pathogenic mutations in HSP, we designed a next generation sequencing-based genetic testing and validation process. To undertake a genetic diagnosis for the family, we conducted quadro whole-exome sequencing on the proband, the two affected children, and the husband to identify the cause of pathogenesis for HSP. We found a novel TUBB4A (NM_006087.4) c.1023C>G (p.F341L). Mutation that was present in the three affected patients but absent in the unaffected individual. Sanger testing of the polymerase chain reaction products detected the c.1023C>G variant in the affected family members, demonstrating the presence of an autosomal dominant pattern (Figure 1B).
- To distinguish whether the variant was an intolerant mutation, the Mutation Taster (www.mutationtaster.org/) tool revealed the evolutionary conservation of altered residues (Figure 1C). The results from Polymorphism Phenotyping (version 2) software (http://genetics.bwh.harvard.edu/pph2/, accessed on June 21, 2021) indicated probable damage affecting protein function. We assessed the frequency of the variants in the general population through datasets including the Exome Aggregation Consortium, dbSNP, and 1000 Genomes Project. The c.1023C>G variant was not detected in these databases. However, the rs368960272 (c.1023C>T) variant was identified, with an allele frequency lower than the usual 0.1% observed in the general and Asian populations. We consider this missense mutation to be a rare variant.
- Using SWISS-MODEL software (Computational Structural Biology Group, Basel, Switzerland;https://www.biozentrum.unibas.ch/research/research-groups/research-groups-a-z/overview/unit/research-group-torsten-schwede ), we discovered that the TUBB4A p.F341L mutation has a high likelihood of causing changes in protein structure (https://swissmodel.expasy.org) (Supplementary Figure 1 in the online-only Data Supplement) [5-7]. DynaMut2 also predicted that the mutation could make the protein structure unstable [8]. We used mCSM–NA (https://biosig.lab.uq.edu.au/mcsm_na/prediction) and mmCSM (https://biosig.lab.uq.edu.au/mmcsm_ppi/) to predict the effects of the TUBB4A p.F341L mutation on protein–protein and protein–DNA interactions. The results showed that the mutation could disrupt these interactions, leading to changes in protein function [8].
- The ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar) included the nearby SNP substitution TUBB4A c.1021T>C, resulting in an identical amino acid change (p.F341L), which has been classified as likely pathogenic. The TUBB4A c.1023C>G (p.F341L) mutation meets the criteria outlined in the American College of Medical Genetics and Genomics (ACMG) guidelines, supporting its classification as likely pathogenic [9]. According to the ACMG guidelines, this mutation satisfies the following criteria: PS1, PM2, PP3, and PP4.
CASE REPORT
- The TUBB4A gene is responsible for making β- and α-tubulins, which are major components of microtubules [10,11]. Microtubules are crucial cytoskeletal structures involved in cell division and intracellular trafficking. Specific isotypes of β–tubulin, such as 2A, 2B, 3, and 4A, are highly abundant neuron-specific proteins in the brain. The mutation in our patients was located at the second globular domain of the TUBB4A gene, specifically in the H10-S9 loop region. Previous reports have identified other mutations, including p.A352T, p.C354Y, p.D355V, and p.M323R, within the H10-S9 loop region of TUBB4A, which all present as TUBB4A spectrum disorders [12-14]. In the 3D structure of tubulin, the p.D355V, pV255I, and p.F341L mutations are located near the intradimer surface, which may cause alterations affecting microtubule dynamics (Supplementary Figure 1 in the online-only Data Supplement). According to previous reports, TUBB4A mutations can cause a range of symptoms, including spasticity, dystonia, chorea, cerebellar ataxia, cognitive impairment, and epilepsy. TUBB4A mutations are usually identified during childhood or infancy, although there have been rare instances of diagnosis in adolescence or adulthood. In Supplementary Table 1 (in the online-only Data Supplement), we compile previous reports detailing cases of isolated hypomyelination and spastic paraplegia. Common manifestations include spasticity and delays in developmental milestones, often accompanied by cognitive impairment. In contrast, our patients displayed a later onset of symptoms and a milder clinical presentation characterized by spasticity, sometimes with subtle cerebellar signs. This clinical phenotype aligns with the definition of complicated HSP. Importantly, our patients exhibited normal cognitive function, and brain MRI scans revealed only minor hypomyelination changes in the cerebral cortex. To our knowledge, this is the first documented case of adult-onset HSP linked to a TUBB4A mutation. In conclusion, the F341L mutation is suspected to be a pathogenic mutation that can manifest as complicated HSP with isolated hypomyelination. The family in our study presented with a late onset and slow progression of spasticity without obvious dystonia or cognitive impairment. As indicated by our findings, clinicians should evaluate the TUBB4A gene in patients presenting with late-onset progressive spastic paraplegia with an autosomal dominant inheritance pattern.
DISCUSSION
Supplementary Material
Supplementary Table 1.
Supplementary Figure 1.
-
Ethics Statement
We obtained informed consent for genetic testing and case reports from the patient, and we performed a genetic analysis to test for hereditary ataxia (IRB No 202200285B0A3 at Chang Gung Memorial Hospital, Linkou Medical Center). The authors certify that this article complies with the Principles of Ethical Publishing of Journal of Movement Disorders and declare that they acted in accordance with ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This work was supported by Minister of Science and Technology, Taiwan (MOST 109-2314-B-182A-077-MY3) and Chang Gung Memorial Hospital, Taoyuan, Taiwan (CMRPG3K1481).
-
Author contributions
Conceptualization: Yih-Ru Wu. Formal analysis: Chih-Ying Chao, Chun-Chieh Wang, Wen-Lang Fan. Funding acquisition: Yih-Ru Wu. Investigation: Yih-Ru Wu, Pei-Chen Hsieh. Methodology: Chih-Ying Chao, Chun-Chieh Wang, Wen-Lang Fan, Yih-Ru Wu. Project administration: Yih-Ru Wu. Resources: Yih-Ru Wu. Software: Pei Shan Yu, Wen-Lang Fan. Supervision: Yih-Ru Wu. Validation: Chih-Ying Chao, Chun-Chieh Wang, Wen-Lang Fan. Visualization: Yih-Ru Wu. Writing—original draft: Pei-Chen Hsieh. Writing—review & editing: Yih-Ru Wu.
Notes

- 1. Sagnelli A, Magri S, Farina L, Chiapparini L, Marotta G, Tonduti D, et al. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and FDG-PET findings. J Neurol 2016;263:591–593.ArticlePubMedPDF
- 2. Curiel J, Rodríguez Bey G, Takanohashi A, Bugiani M, Fu X, Wolf NI, et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum Mol Genet 2017;26:4506–4518.ArticlePubMedPMCPDF
- 3. Blumkin L, Halevy A, Ben-Ami-Raichman D, Dahari D, Haviv A, Sarit C, et al. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics 2014;15:107–113.ArticlePubMedPDF
- 4. Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L, et al. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov Disord 2015;30:854–858.ArticlePubMedPDF
- 5. Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997;18:2714–2723.ArticlePubMed
- 6. Peitsch MC. ProMod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem Soc Trans 1996;24:274–279.ArticlePubMedPDF
- 7. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods 2022;19:679–682.ArticlePubMedPMCPDF
- 8. Rodrigues CHM, Pires DEV, Ascher DB. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci 2021;30:60–69.PubMed
- 9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med 2015;17:405–424.ArticlePubMedPMCPDF
- 10. Romaniello R, Arrigoni F, Fry AE, Bassi MT, Rees MI, Borgatti R, et al. Tubulin genes and malformations of cortical development. Eur J Med Genet 2018;61:744–754.ArticlePubMed
- 11. Simons C, Wolf Nicole I, McNeil N, Caldovic L, Devaney JM, Takanohashi A, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the Leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013;92:767–773.ArticlePubMedPMC
- 12. Carvalho D, Santos S, Martins B, Pinto Marques F. TUBB4A novel mutation reinforces the genotype–phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum. Brain 2015;138(Pt 2):e327.ArticlePubMed
- 13. Lu Y, Ondo Y, Shimojima K, Osaka H, Yamamoto T. A novel TUBB4A mutation G96R identified in a patient with hypomyelinating leukodystrophy onset beyond adolescence. Hum Genome Var 2017;4:17035.ArticlePubMedPMCPDF
- 14. Di Bella D, Magri S, Benzoni C, Farina L, Maccagnano C, Sarto E, et al. Hypomyelinating leukodystrophies in adults: clinical and genetic features. Eur J Neurol 2021;28:934–944.ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite