E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 17(1); 2024 > Article
-
Letter to the editor
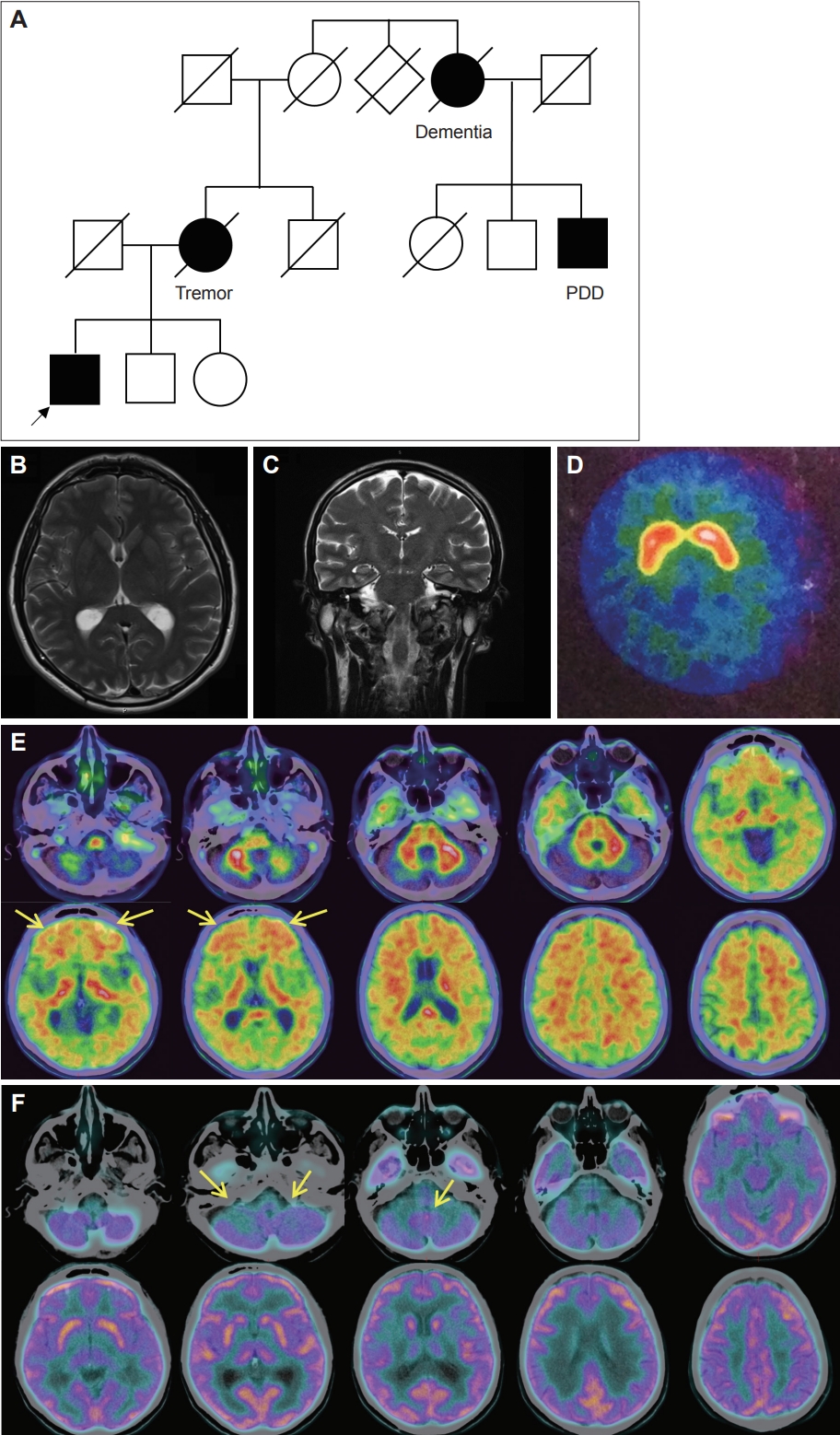
A Novel Pathogenic PSEN1 Variant in a Patient With Dystonia-Parkinsonism Without Dementia -
Maria Chiara Malaguti1

 , Alessio Di Fonzo2, Chiara Longo1,3, Raffaella Di Giacopo4, Costanza Papagno5, Davide Donner6,7, Umberto Rozzanigo8, Edoardo Monfrini2,9
, Alessio Di Fonzo2, Chiara Longo1,3, Raffaella Di Giacopo4, Costanza Papagno5, Davide Donner6,7, Umberto Rozzanigo8, Edoardo Monfrini2,9 -
Journal of Movement Disorders 2024;17(1):102-105.
DOI: https://doi.org/10.14802/jmd.23125
Published online: September 14, 2023
1Department of Neurology, Santa Chiara Hospital, APSS, Trento, Italy
2Department of Neurology, Foundation Istituti di Ricovero e Cura a Carattere Scientifico Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
3Department of Psychology, University of Milano-Bicocca, Milan, Italy
4Department of Neurology, Rovereto Hospital, APSS, Rovereto, Italy
5Center for Mind/Brain Sciences (CIMeC), University of Trento, Rovereto, Italy
6Department of Nuclear Medicine, Santa Chiara Hospital, APSS, Trento, Italy
7Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
8Department of Diagnostic Imaging, Santa Chiara Hospital, APSS, Trento, Italy
9Department of Pathophysiology and Transplantation, Neuroscience Section, Dino Ferrari Center, University of Milan, Milan, Italy
- Corresponding author: Maria Chiara Malaguti, MD Department of Neurology, Santa Chiara Hospital, APSS, Largo Medaglie d’Oro, 9, Trento 38122, Italy / Tel: +39-0461-903281 / Fax: +39-0461-902732 / E-mail: mariachiara.malaguti@apss.tn.it
Copyright © 2024 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 877 Views
- 62 Download
Supplementary Material
Video 1.
Supplemental Table 1.
-
Ethics Statement
The authors confirm that the approval of an Institutional Review Board (IRB) was not required for this work. Informed consent was obtained from the patient for genetic testing and case report. The authors certify that this article complies with the Principles of Ethical Publishing of Journal of Movement Disorders and declare that they acted in accordance with ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None
-
Author contributions
Conceptualization: Maria Chiara Malaguti, Alessio Di Fonzo, Edoardo Monfrini. Project administration: Maria Chiara Malaguti, Alessio Di Fonzo, Edoardo Monfrini. Data curation: Maria Chiara Malaguti, Alessio Di Fonzo, Chiara Longo, Edoardo Monfrini. Investigation: all authors. Methodology: Maria Chiara Malaguti, Alessio Di Fonzo, Edoardo Monfrini. Visualization: Maria Chiara Malaguti, Alessio Di Fonzo, Edoardo Monfrini. Writing—original draft: Maria Chiara Malaguti, Chiara Longo, Alessio Di Fonzo, Edoardo Monfrini. Writing—review & critique: all authors.
Notes
- 1. Jayne T, Newman M, Verdile G, Sutherland G, Münch G, Musgrave I, et al. Evidence for and against a pathogenic role of reduced γ-secretase activity in familial Alzheimer’s disease. J Alzheimers Dis 2016;52:781–799.ArticlePubMed
- 2. Takao M, Ghetti B, Hayakawa I, Ikeda E, Fukuuchi Y, Miravalle L, et al. A novel mutation (G217D) in the Presenilin 1 gene (PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol 2002;104:155–170.ArticlePubMedPDF
- 3. Winkel I, Ermann N, Żelwetro A, Sambor B, Mroczko B, Kornhuber J, et al. Cerebrospinal fluid α synuclein concentrations in patients with positive AD biomarkers and extrapyramidal symptoms. J Neural Transm (Vienna) 2021;128:817–825.ArticlePubMedPMCPDF
- 4. Bagaria J, Bagyinszky E, An SSA. Genetics, functions, and clinical impact of presenilin-1 (PSEN1) gene. Int J Mol Sci 2022;23:10970.ArticlePubMedPMC
- 5. Zhang S, Lei C, Liu P, Zhang M, Tao W, Liu H, et al. Association between variant amyloid deposits and motor deficits in FAD-associated presenilin-1 mutations: a systematic review. Neurosci Biobehav Rev 2015;56:180–192.ArticlePubMed
- 6. Carecchio M, Picillo M, Valletta L, Elia AE, Haack TB, Cozzolino A, et al. Rare causes of early-onset dystonia-parkinsonism with cognitive impairment: a de novo PSEN-1 mutation. Neurogenetics 2017;18:175–178.ArticlePubMedPDF
- 7. Gatto EM, Rojas GJ, Nemirovsky SI, Da Prat G, Persi G, Cesarini M, et al. A novel mutation in PSEN1 (p.Arg41Ser) in an Argentinian woman with early onset parkinsonism. Parkinsonism Relat Disord 2020;77:21–25.ArticlePubMed
- 8. Chen Y, Liu P, Xie F, Wang B, Lin Z, Luo W. A heterozygous de novo PSEN1 mutation in a patient with early-onset parkinsonism. Neurol Sci 2022;43:1405–1409.ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite