E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(1); 2023 > Article
-
Review Article
Multiple System Atrophy: Advances in Diagnosis and Therapy -
Hirohisa Watanabe1

 , Sayuri Shima1, Yasuaki Mizutani1, Akihiro Ueda1,2, Mizuki Ito1,3
, Sayuri Shima1, Yasuaki Mizutani1, Akihiro Ueda1,2, Mizuki Ito1,3 -
Journal of Movement Disorders 2023;16(1):13-21.
DOI: https://doi.org/10.14802/jmd.22082
Published online: December 20, 2022
1Department of Neurology, Fujita Health University, School of Medicine, Toyoake, Japan
2Department of Neurology, Fujita Health University Okazaki Medical Center, Okazaki, Japan
3Department of Neurology, Fujita Health University Bantane Hospital, Nagoya, Japan
- Corresponding author: Hirohisa Watanabe, MD, PhD Department of Neurology, Fujita Health University, School of Medicine, 1-98 Dengakugakubo, Kutsukake-cho, Toyoake 470-1192, Japan / Tel: +81- 562-93-9295 / Fax: +81-562-93-1856 / E-mail: hirohisa.watanabe@gmail.com
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- This review summarizes improvements in understanding the pathophysiology and early clinical symptoms of multiple system atrophy (MSA) and advancements in diagnostic methods and disease-modifying therapies for the condition. In 2022, the Movement Disorder Society proposed new diagnostic criteria to develop disease-modifying therapies and promote clinical trials of MSA since the second consensus was proposed in 2008. Regarding pathogenesis, cutting-edge findings have accumulated on the interactions of α-synuclein, neuroinflammation, and oligodendroglia with neurons. In neuroimaging, introducing artificial intelligence, machine learning, and deep learning has notably improved diagnostic accuracy and individual analyses. Advancements in treatment have also been achieved, including immunotherapy therapy against α-synuclein and serotonin-targeted and mesenchymal stem cell therapies, which are thought to affect several aspects of the disease, including neuroinflammation. The accelerated progress in clarifying the pathogenesis of MSA over the past few years and the development of diagnostic techniques for detecting early-stage MSA are expected to facilitate the development of disease-modifying therapies for one of the most intractable neurodegenerative diseases.
- Multiple system atrophy (MSA) is a progressive neurodegenerative disease characterized by varying degrees of autonomic failure (AF), parkinsonism, and cerebellar ataxia [1]. Pathologically, it is characterized by glial cytoplasmic inclusions (GCIs), which consist of insoluble α-synuclein accumulated within oligodendrocytes. MSA is classified as an α-synucleinopathy along with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB).
- Cutting-edge technology has clarified the structure of α-synuclein in MSA, and pathophysiological and differential diagnostic techniques have been developed based on structural differences [2,3]. Additionally, increasing evidence supports the involvement of neuroinflammation and oligodendroglial cell changes in MSA pathogenesis [4-8].
- Symptomatic treatment has limited efficacy in MSA, and no treatments suppress or alleviate disease progression. However, notable advancements have been achieved in the development of antibody therapies against α-synuclein [9], multitargeted disease-modifying therapies, including mesenchymal stem cell (MSC)-mediated neuroinflammation treatments, and other symptomatic treatments, such as selective serotonin reuptake inhibitors (SSRIs) [10-14].
- In 2022, the Movement Disorder Society (MDS) proposed new diagnostic criteria to develop disease-modifying therapies and promote clinical trials of MSA [15], which was the first time that it had been revised since the second consensus was proposed in 2008. Additionally, novel biomarkers for diagnosis and progression have been identified from brain imaging, and blood and cerebrospinal fluid (CSF) data have been reported [16-26].
- This review summarizes recent developments in MSA research, clarifying the early pathophysiology, psychiatric and cognitive symptoms, diagnostic biomarker development, and drug discovery.
INTRODUCTION
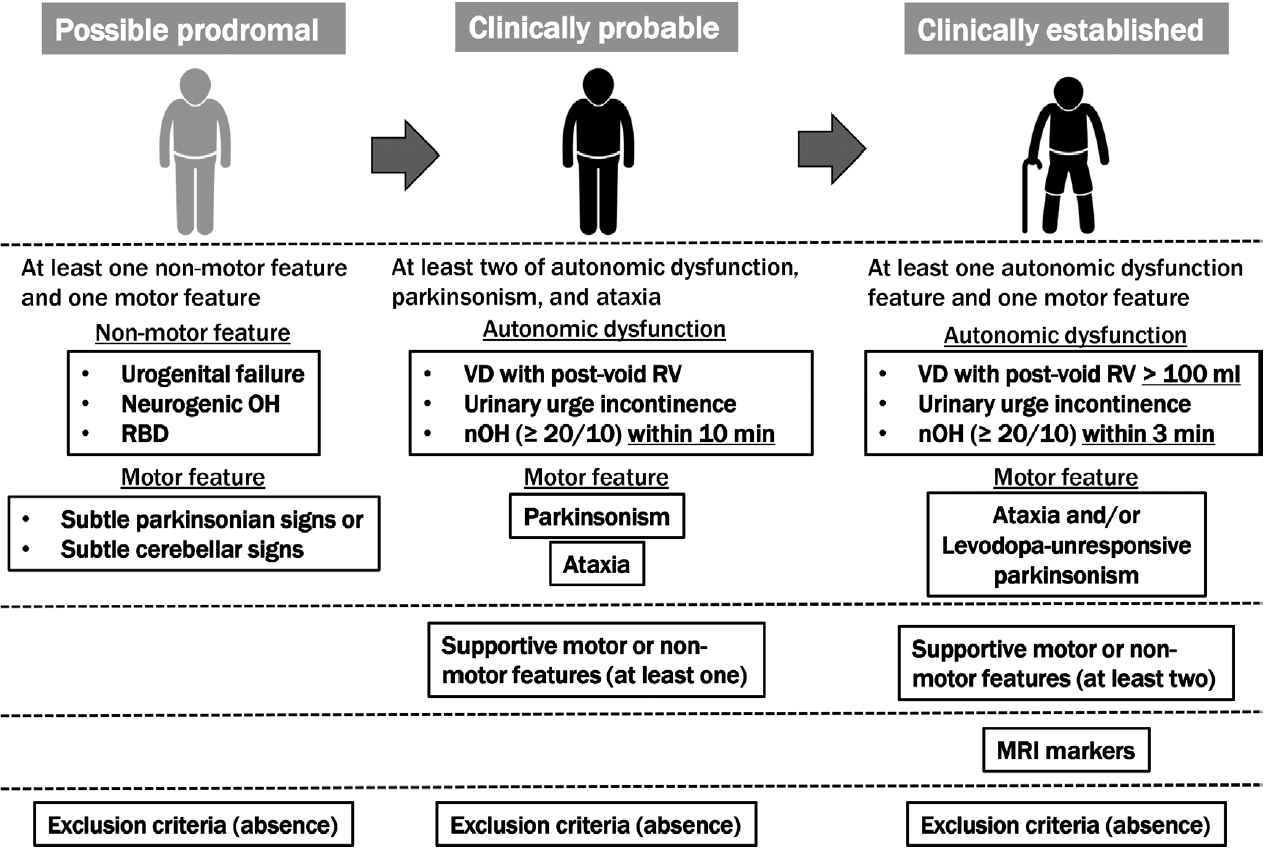
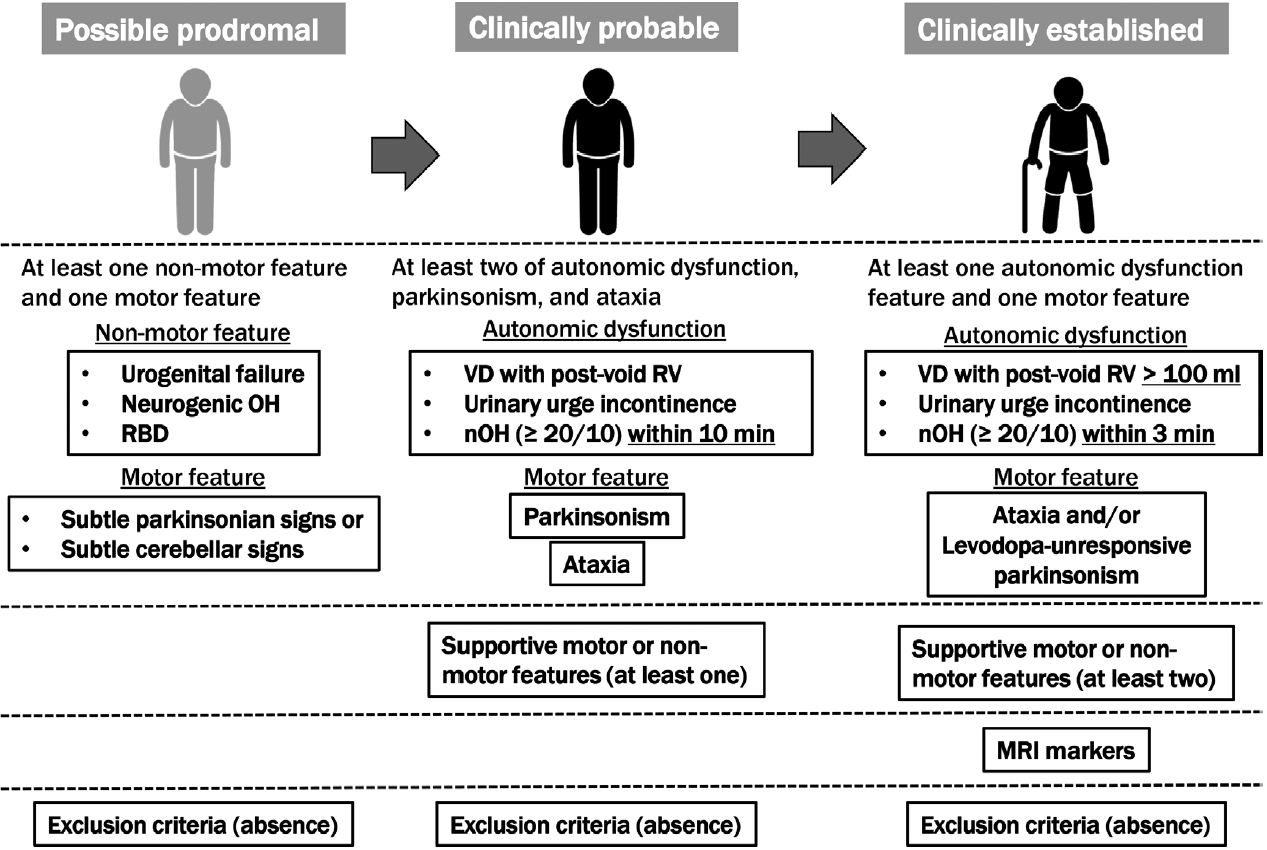
- The second consensus criteria, which are widely used to diagnose MSA, were not sufficiently sensitive for the early stages; therefore, since many patients with MSA have been excluded from clinical trials for disease-modifying therapy, the MDS developed the current criteria for MSA diagnosis to improve accuracy early in the course of the disease. According to the MDS criteria, MSA is classified into four levels of certainty: neuropathologically established MSA, clinically established MSA, clinically probable MSA, and possible prodromal MSA.
- The second consensus criterion was consistent with the neuropathologically established MSA: findings of widespread and abundant α-synuclein-positive GCIs in the central nervous system associated with neurodegenerative changes in striatonigral or olivopontocerebellar structures that characterize MSA on autopsy. A distinction between the MSA-parkinsonian type (MSA-P) and MSA-cerebellar type (MSA-C) is evident according to the predominant motor phenotype. The proposed new category of possible prodromal MSA with very low specificity is expected to undergo continuous refinement with emerging data, particularly from prospective investigations and biomarker studies. All three clinical diagnostic categories (clinically established, clinically probable, and possible prodromal MSAs) need validation in future studies. For clinically established MSA, clinically probable and possible prodromal MSA, symptoms must begin after 30 years of age because no MSA cases have been confirmed postmortem in the third decade of life or earlier; no past family history can be present, and the disease must progress, as in the second consensus criteria. Older-onset MSA has been clinically and pathologically demonstrated, and MSA-P has been reported to be a significant disease type in South Korea and Japan [27]. Patients with MSA onset after 75 years of age were excluded from the second consensus statement but not from the MDS MSA criteria. Figure 1 summarizes the concept of the MDS criteria for the clinical diagnosis of MSA. Table 1 compares supportive motor and nonmotor features, magnetic resonance imaging (MRI) markers of clinically established MSA, exclusion criteria among MDS criteria, and additional features of possible MSA, supportive features, and nonsupporting features in the second consensus statement.
- Clinically established MSA is defined as a combination of 1) core clinical features (at least one of three AFs and poorly levodopa-responsive parkinsonism and/or cerebellar syndrome), 2) at least two supportive motor or nonmotor features, 3) at least one brain MRI marker suggestive of MSA, and 4) no exclusion criteria met. The severity and definition of orthostatic hypotension differed from those in the second consensus criteria. In the MDS criteria, supportive features and MRI markers are necessary for clinically established MSA compared to the second consensus statement. Rapid progression, moderate-to-severe postural instability, severe speech impairment and dysphagia within three years of motor onset; craniocervical dystonia induced or exacerbated by levodopa in the absence of limb dyskinesia; unexplained Babinski signs; jerky myoclonic postural or kinetic tremors; and postural deformities were the eight motor supportive features. Stridor, inspiratory signs, cold, discolored hands and feet, erectile dysfunction (in those aged < 60 years for clinically probable MSA), and pathological laughter or crying were the five nonmotor features. MRI markers of clinically established MSA-P include atrophy of the putamen (and signal decreases on iron-sensitive sequences), middle cerebellar peduncle, pons, or cerebellum, the hot-cross-bun sign, and increased diffusivity of the putamen or middle cerebellar peduncle. MRI markers of clinically established MSA-C included atrophy of the putamen (and signal decreases on iron-sensitive sequences), middle cerebellar peduncle, or pons, the hot-cross-bun sign, and increased diffusivity of the putamen.
- Clinically probable MSA was defined as a combination of 1) AF, parkinsonism, and cerebellar impairment; at least two in any combination, including parkinsonism combined with cerebellar impairment without AF, although AF was essential in the second consensus statement, 2) at least one supportive feature, and 3) no exclusion criteria met. The orthostatic hypotension and residual volume definitions for clinically probable MSA differed from those for clinically established MSA (Figure 1).
- The exclusion criteria for clinically established and clinically probable MSA included 1) substantial and persistent beneficial response to dopaminergic medications, 2) unexplained anosmia on olfactory testing, 3) fluctuating cognition with pronounced variation in attention and alertness and early decline in visuoperceptual abilities, 4) recurrent visual hallucinations not induced by drugs within three years of disease onset, 5) dementia according to Diagnostic & Statistical Manual of Mental Disorders (DSM)-V within three years of disease onset, 6) downgaze supranuclear palsy or slowing of vertical saccades, 7) brain MRI findings suggestive of an alternative diagnosis (e.g., progressive supranuclear palsy [PSP], multiple sclerosis, vascular parkinsonism, symptomatic cerebellar disease), and 8) documentation of an alternative condition (MSA-like condition, including genetic or symptomatic ataxia and parkinsonism) known to produce AF, ataxia, or parkinsonism and plausibly connected to the patient’s symptoms.
- Possible prodromal MSA is defined as a combination of 1) at least one of three clinical nonmotor features as entry criteria (polysomnography-proven rapid eye movement [REM] sleep behavior disorder, neurogenic orthostatic hypotension [decrease ≥ 20/10 mm Hg in blood pressure] within 10 min of standing or head-up tilt, urogenital failure [erectile dysfunction in men aged < 60 years combined with unexplained voiding difficulties with postvoid urinary residual volume > 100 mL or unexplained urinary urge incontinence]), 2) at least one of the subtle parkinsonian or cerebellar signs, and 3) absence of exclusion criteria. Exclusion criteria for possible prodromal MSA included the following: unexplained anosmia in olfactory testing and/or abnormal cardiac sympathetic imaging (123I-metaiodobenzylguanidine-scintigraphy); fluctuating cognition with pronounced variation in attention and alertness and early decline in visuoperceptual abilities; recurrent visual hallucinations not induced by drugs within three years of disease onset; dementia according to DSM-V within three years of disease onset; downgaze supranuclear gaze palsy or slowing vertical saccades; brain MRI findings suggestive of an alternative diagnosis (e.g., PSP, multiple sclerosis, vascular parkinsonism, symptomatic cerebellar disease, etc.); and documentation of an alternative condition (MSA-like condition, including genetic or symptomatic ataxia and parkinsonism) known to produce AF, ataxia, or parkinsonism and plausibly connected to the patient’s symptoms. More recently, novel biomarkers for MSA diagnosis have been provided but were excluded from the new MDS criteria given the limited availability, suboptimal diagnostic accuracy, or lack of diagnostic validation. However, a biomarker, which will allow the improved diagnosis of MSA, will be applied to all MSA diagnostic categories.
MOVEMENT DISORDER SOCIETY CRITERIA FOR THE DIAGNOSIS OF MULTIPLE SYSTEM ATROPHY
- The clinical diagnosis of MSA requires the presence of various combinations of AF and/or parkinsonism or cerebellar ataxia [1,15]. However, the median time from disease onset that meets the criteria for probable diagnosis based on the second consensus criteria is approximately two years [28]. Moreover, patients often have only AF, parkinsonism, or cerebellar ataxia early during the disease [29]. Therefore, in addition to the novel MDS diagnostic criteria, a biomarker that can diagnose this stage of “monosystem atrophy” with high accuracy is desirable.
- In a recent report reviewing clinical symptoms before MSA diagnosis using a sizable medical database [30], hypotension, urinary disturbance, dizziness, and disequilibrium were more frequent. Additionally, the importance of new-onset or exacerbated snoring has also been noted [31]. These clinical indicators, however, are less specific because they can be detected in the presence of diseases other than MSA or the absence of organic disease.
- Concerning cognitive dysfunction and psychiatric symptoms, memory impairment and depression have also been reported as prodromal symptoms of MSA with high frequencies [32]. In particular, a history of depression was confirmed more than five years before diagnosis. In a study, following the onset of MSA, 40% of patients had mild to moderate cognitive dysfunction with or without orthostatic hypotension, which was characterized by executive dysfunction and verbal memory impairment, with depression in 28% and anxiety disorders in 22% of patients [32]. According to a neuropathological study, neurocytoplasmic inclusions in the hippocampal dentate gyrus, Ammon’s horn regions [33], and the entorhinal cortex may cause memory deterioration. Investigation of the pathogenesis of cognitive dysfunction, including complex pathology, remains a future challenge.
OTHER EARLY CLINICAL PHENOTYPES AND FEATURES
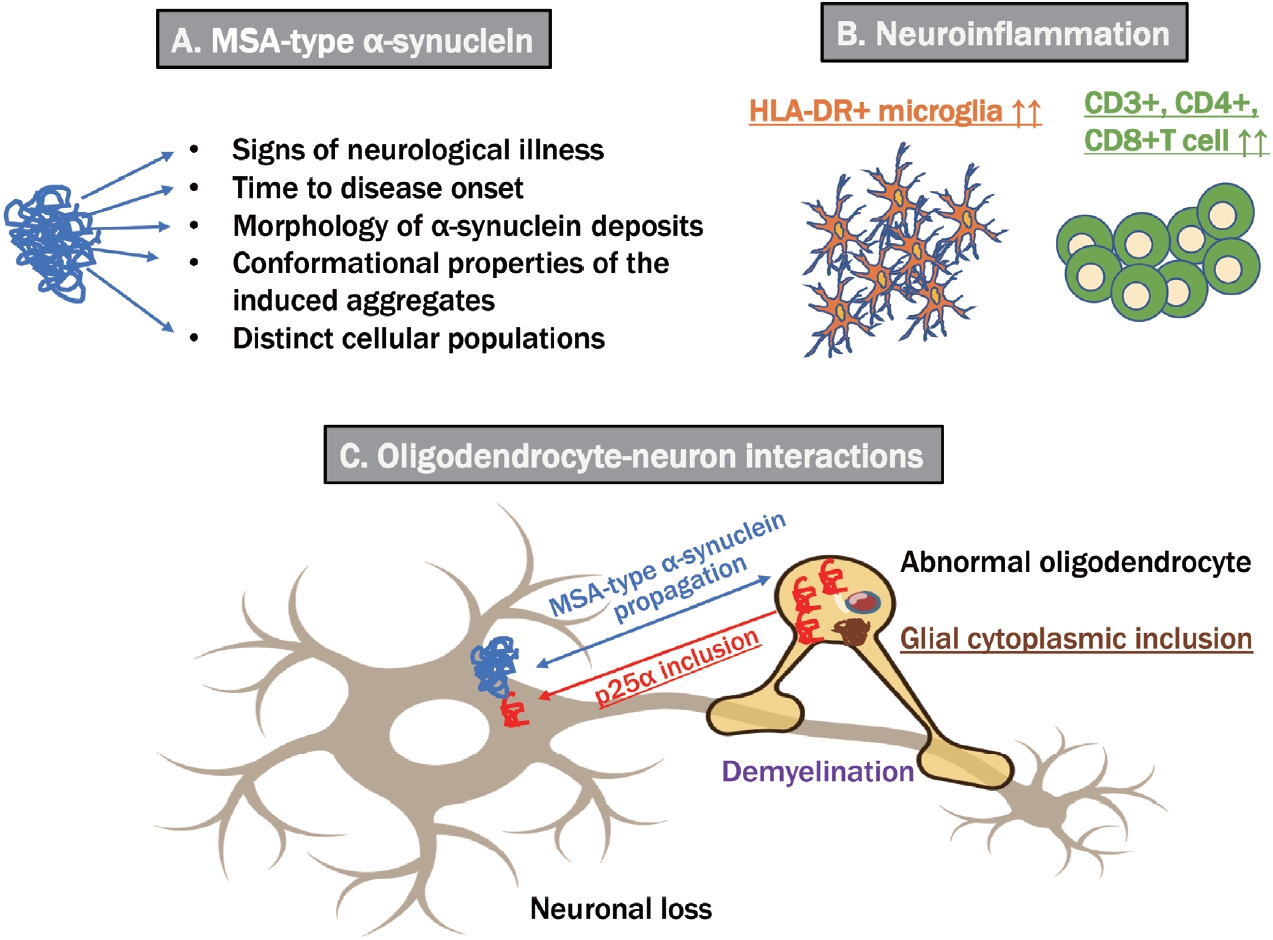
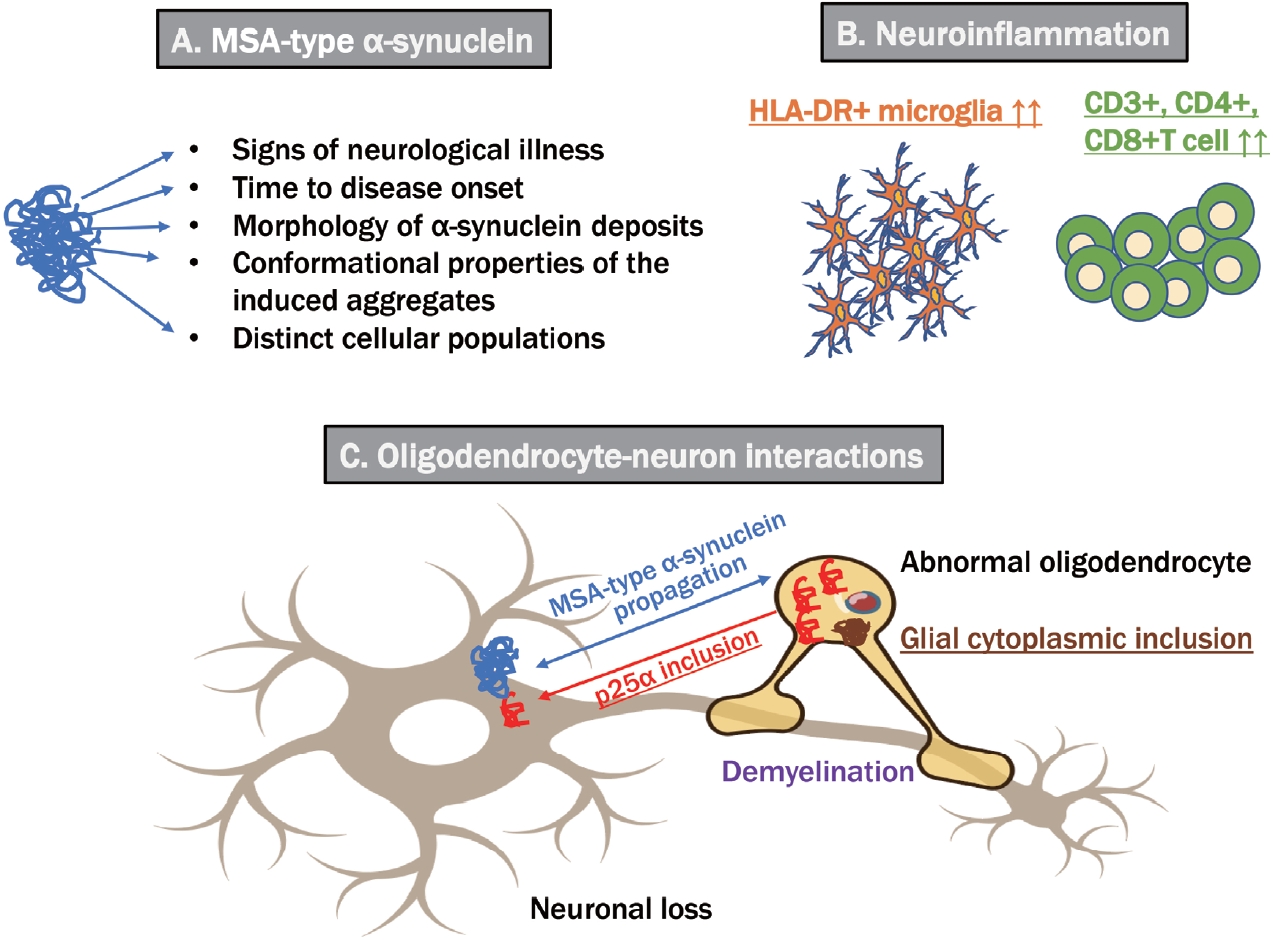
- Synucleinopathies, which are divided into Lewy body diseases, including PD, DLB, and MSA, are clinically and pathologically heterogeneous disorders characterized by pathological aggregates of α-synuclein in neurons and glia in the form of Lewy bodies and neurites, neuronal cytoplasmic inclusions, and GCIs. Recent breakthroughs have demonstrated that α-synuclein species derived from Lewy body disease and MSA are distinct “strains” with different seeding properties. First, cryo-electron microscopy shows the following characteristic structures of α-synuclein in the brains of patients with MSA: 1) α-synuclein consists of two different protofilaments, 2) the filaments of α-synuclein from the brains of patients with MSA and DLB are different, and 3) the structure of α-synuclein filaments extracted from the brains of patients with MSA was found to differ from the structure formed in vitro using recombinant proteins [2]. A study in which genetically modified mice were inoculated with recombinant or brainderived α-synuclein aggregates with different structures showed that structural differences might cause differences in clinical presentation, the time to disease onset, and the structure of α-synuclein brain deposits, in addition to the types of cells deposited and α-synuclein propagation [3]. GCIs from patients with MSA injected into the striatum of nonhuman primates resulted in the loss of substantia nigra dopaminergic neurons and medium-sized striatal spinous cells and the loss of oligodendrocytes in the same region, and demyelination, neuroinflammation, and α-synuclein pathology were noted [34]. Despite controversy regarding oligodendrocytes’ α-synuclein expression, MSA-derived extracts can exacerbate oligodendroglial α-synuclein pathology. Furthermore, nigral and striatal degeneration appears to precede GCI formation, which is linked to a-synuclein redistribution in oligodendrocytes. However, Lewy bodies injected into nonhuman primates did not show such redistribution [34]. As a result, disease-related strains could differentiate between PD and MSA. These findings indicated the significant role of structural abnormalities in α-synuclein in the pathogenesis of MSA (Figure 2).
- The role of inflammatory pathogenesis has also offered important insights, with significant increases in HLA-DR+ microglia in the putamen and substantia nigra of patients with MSA compared with controls and significant increases in CD3+, CD4+, and CD8+ T cells in these brain regions [4]. Furthermore, [11C]PBR28 positron emission tomography (PET), which visualizes the translocator protein expressed in glial cells, revealed a significant increase in the accumulation of lentiform nuclei and cerebellar white matter in MSA compared with PD, and both diseases could be differentiated with 83% sensitivity and 100% specificity [5]. Conversely, a genome-wide association study revealed a multifactorial overlap between MSA and inflammatory bowel disease, identifying three shared loci with reading variants upstream of the DENND1B and RSP04 genes and in an intron of the C7 gene, which has been implicated in various neuroinflammatory conditions [6].
- GCI, a diagnostic indicator of MSA, is generated within oligodendroglia. p25α/tubulin polymerization-promoting protein is expressed in mature oligodendroglia and is essential for myelin reorganization and stabilization. Ferreira et al. [7] found that in MSA, 1) oligodendroglial cell body expansion and myelin primary protein degradation occur; 2) p25α/TPP leaves the nucleus and forms an inclusion body in the cytoplasm; 3) the α-synuclein monomer enters the inclusion body, and p25α/TPP becomes nucleated and forms MSA-type α-synuclein aggregates; and 4) p25α/TPP can be released from oligodendroglial cells to form nuclear and cytoplasmic inclusions in neurons. Moreover, a study profiling the expression of microRNAs in the serum of 20 MSA cases compared with 40 controls via genome-wide analysis found 25 microRNAs associated with MSA [8]. In particular, the expression of microRNA-7641, which regulates axonal myelination, and microRNA-191, which is involved in T-cell survival and prion pathways, were significantly different between MSA and PD, which is worth noting considering the association between neuroinflammation and abnormal oligodendroglia.
ADVANCES IN UNDERSTANDING THE PATHOPHYSIOLOGY
- New diagnostic methods focusing on MSA-type α-synuclein are being developed. The properties of α-synuclein aggregates in CSF treated with protein misfolding cyclic amplification (PMCA) and real-time Quaking-Induced Conversion (RT-QuIC) provided useful findings for the early diagnosis of MSA and differentiation from PD. Initially, the PMCA reaction was performed by cyclic sonication and incubating infected samples with typical brain homogenates containing cellular prion protein (PrPC) as the substrate. Then, PMCA products were analyzed with protease digestion, and Western blotting was performed to detect amplified conversion products. However, as a limitation, PMCA requires a week-long reaction time, and Western blotting carries the risk of biohazardous infectivity of the amplified products. In contrast, RT-QuIC provides much shorter assay times (e.g., 1–2 days) and noninfectious amplification products using multiwell plates; shaking instead of sonication, a recombinant rather than brain-derived PrPC substrate, and fluorescence (thioflavin T) instead of a Western blot readout (Figure 3).
- Further development of PMCA and RT-QuIC assays has improved their sensitivity, specificity and applicability to most prion diseases and proteinopathies. PD/DLB and MSA can be differentiated with high sensitivity and specificity using the differences in their aggregation rate and intensity of maximum fluorescence [16]. Interestingly, RT-QuIC showed high seeding activity in CSF from patients with PD, DLB, pure AF, and idiopathic REM sleep behavior disorders but no increase in seeding activity in those with MSA [17]. However, the results on RT-QuIC in MSA are not consistent among studies, and some studies report positive RT-QuIC in MSA [18,19]. Although no study has directly compared PMCA and RT-QuIC, differences in the structure of α-synuclein may have influenced the results, and further investigation is warranted. Concerning the immunostaining results for α-synuclein phosphorylated explicitly at serine 129 residues in nerves in the skin of the neck, thighs, and lower legs, 72% of patients with MSA-P showed positive findings, mainly in somatic fibers in the distal subepidermal plexus. In contrast, all patients with PD and orthostatic hypotension had positive findings of autonomic cutaneous fibers in the proximal and distal areas [20]. Differences in the structure of α-synuclein may also influence these differences in sites with lesion tendencies.
- The neurofilament light chain (NFL), a cytoskeleton component abundant in the axonal projections of neurons, can be elevated in CSF and blood, indicating neuronal damage. NFL in CSF is significantly elevated in MSA compared to PD and DLB, and α-synuclein measured using PMCA was characterized by faster aggregation but lowered maximal fluorescence in MSA compared to PD/DLB [21]. By combining the results of NFL and PMCA-based α-synuclein, differentiating MSA from PD/DLB with high sensitivity and specificity may be possible. NFL in blood also correlates well with the unified MSA rating scale (UMSARS) and is expected to serve as a biomarker of progression [22].
- For novel imaging biomarkers, diffusion MRI was reported to detect abnormalities of the middle cerebellar peduncle and putamen in MSA with notable sensitivity. However, given that most studies used a manual region of interest (ROI), discrepancies in results have been a problem. Krismer et al. [23] reported that combining automated ROIs with machine learning and mean diffusivity, a measure of diffusion MRI in the middle cerebellar peduncle and putamen, can be a good discriminator between early and mid-stage MSA and PD. In a study of 1,002 patients with MSA at 17 centers, machine learning was used to analyze data from 60 template regions and fiber bundles on MRI diffusionweighted images, validating the discriminative ability for PD, MSA, and PSP. In this study, the area under the curve (AUC) for differentiating PD from MSA/PSP was 0.955, and the AUC for differentiating MSA from PSP was 0.926 [24]. Voxel-based morphometry, a method for standardizing differences between facilities compared to diffusion MRI, usually requires group comparisons, but individual analysis has recently become possible. Ebina et al. [25] applied a generalized linear model to each voxel of brain volume using age, sex, and total brain volume as covariates, which affected the results of individual analyses. They observed high diagnostic accuracy (individual voxel-based morphometry adjusting covariates) for differentiating MSA from PD. The combination of automation and machine learning will continue improving diagnostic accuracy and standardizing methods for individual analyses.
- In a study focusing on laryngeal findings, 43.9% of patients with MSA showed clinically evident laryngeal dysfunction and inspiratory stridor. Endoscopic observation of laryngeal movement revealed abnormal findings in 93% of patients with MSA compared with only 1.8% of patients with PD (p < 0.0001). Additionally, irregular laryngeal cartilage movement was observed in 91.2% of patients with MSA but not in patients with PD (p < 0.0001). Various findings were also noted, including impaired vocal fold movement (75.4%), paradoxical vocal fold movement (33.3%), and vocal fold fixation (19.3%), signifying that laryngeal findings are diagnostic indicators of MSA [26].
DIAGNOSTIC BIOMARKER DEVELOPMENT
- Novel disease-modifying therapies for MSA are being developed to inhibit α-synuclein aggregation, alleviate neuroinflammation and confer neuroprotective effects [35]. In a study of PD01A and PD03A (α-synuclein peptide vaccines) administered to 30 patients with MSA, no safety or tolerability issues were observed in either group [9]. Rapid and sustained antibody production targeting α-synuclein was obtained in the PD01A group, but no significant clinical scores were observed during the observation period. Epigallocatechin gallate, a polyphenolic constituent of green tea that inhibits α-synuclein aggregation in basic research, did not significantly improve the UMSARS scores [36].
- Extracellular α-synuclein aggregates can induce neurotoxic signals via neuroinflammation through microglial activation and the release of inflammatory factors. Autologous MSCs are expected to modulate neuroinflammation, inhibit cell death and intercellular transmission, promote neurogenesis and autophagy, degrade α-synuclein aggregates, stabilize axonal transport and phagocytose α-synuclein by microglia [10]. Singer et al. [11] found that the progression rate (UMSARS total scores) among 24 patients with MSA treated with intrathecal adipose-derived MSCs was significantly lower than that among matched historical controls (0.40 ± 0.59 vs. 1.44 ± 1.42 points/month, p = 0.004), with a clear dose-dependent effect. In 2012, Lee et al. [12] reported the potential of MSC therapy for MSA. Further development of MSC therapy is essential.
- Other treatments, such as the mammalian target of rapamycin inhibitor sirolimus, which induces autophagy, were ineffective after 48 weeks of treatment [37]. A natural history study showed that low vitamin B12 levels (< 367 ng/L) were associated with shorter survival (hazard ratio, 1.8; 95% confidence interval, 1.3–2.7), increased frequency of falls within three years of onset, and a lower body mass index, suggesting a potential new target for therapeutic intervention [14].
- Another retrospective natural history study revealed that patients with MSA treated with SSRIs showed a better prognosis than untreated patients [38]. Fluoxetine, a second-generation antidepressant categorized as an SSRI, did not affect the primary endpoint. However, it significantly improved UMSARS scores in Part II (motor examination) and quality-of-life scores related to emotional and social concerns at 12 weeks compared with baseline [13]. Brain 5-HT1A receptor-binding PET showed that 5-hydroxytryptamine dysfunction in several brain regions in MSA might contribute to fatigue, pain, and apathy [39]. Patients with autopsy-confirmed MSA with AFs in the absence of motor symptoms/signs preferentially showed the involvement of medullary serotonergic neurons and autonomic systems in the occurrence of sudden death [40]. Further studies are needed to determine whether serotonin-targeted therapy alleviates several symptoms and modifies the disease course of MSA.
DISEASE-MODIFYING THERAPIES AND OTHER TREATMENT
- Considerable progress in elucidating the structure of α-synuclein in MSA has offered novel insights into MSA pathophysiology and the development of breakthrough diagnostic tools. Neuroinflammation and oligodendroglia changes contribute to MSA pathogenesis. Introducing new methods, including artificial intelligence and machine and deep learning, has resulted in advancements in existing brain imaging tools. Moreover, clinical trials of immunotherapy against α-synuclein, multitargeted disease-modifying therapies, including MSC therapies, and various symptomatic therapies have provided new perspectives. Therefore, the development of MDS diagnostic criteria, early diagnostic methods, and cutting-edge disease-modifying treatments based on advances in understanding the pathophysiology of MSA is expected.
CONCLUSION
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This work was supported by Grants-in-Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (20317603).
-
Author contributions
Conceptualization: Hirohisa Watanabe. Data curation: Yasuaki Mizutani, Mizuki Ito. Formal analysis: Hirohisa Watanabe. Funding acquisition: Hirohisa Watanabe. Investigation: Hirohisa Watanabe. Methodology: Hirohisa Watanabe. Supervision: Akihiro Ueda. Validation: Sayuri Shima. Visualization: Hirohisa Watanabe. Writing—original draft: Hirohisa Watanabe. Writing—review & editing: Sayuri Shima, Yasuaki Mizutani, Akihiro Ueda, Mizuki Ito.
Notes

| MDS criteria for the diagnosis of MSA | Second consensus statement on the diagnosis of MSA | ||||
|---|---|---|---|---|---|
| Supportive motor features | Additional features of possible MSA | ||||
| 1. Rapid progression within 3 years of motor onset | For possible MSA-P or MSA-C | ||||
| 2. Moderate to severe postural instability within 3 years of motor onset | 1. Babinski sign with hyperreflexia | ||||
| 3. Craniocervical dystonia induced or exacerbated by L-dopa in the absence of limb dyskinesia | 2. Stridor | ||||
| 4. Severe speech impairment within 3 years of motor onset | For possible MSA-P | ||||
| 5. Severe dysphagia within 3 years of motor onset | 1. Rapidly progressive parkinsonism | ||||
| 6. Unexplained Babinski sign | 2. Poor response to levodopa | ||||
| 7. Jerky myoclonic postural or kinetic tremor | 3. Postural instability within 3 years of motor onset | ||||
| 8. Postural deformities | 4. Gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction | ||||
| Supportive nonmotor features | 5. Dysphagia within 5 years of motor onset | ||||
| 1. Stridor | 6. Atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum | ||||
| 2. Inspiratory sighs | 7. Hypometabolism on FDG-PET in putamen, brainstem, or cerebellum* | ||||
| 3. Cold, discolored hands and feet | For possible MSA-C | ||||
| 4. Erectile dysfunction (below 60 years of age for clinically probable MSA) | 1. Parkinsonism (bradykinesia and rigidity) | ||||
| 5. Pathologic laughter or crying | 2. Atrophy on MRI of the putamen, middle cerebellar peduncle, or pons | ||||
| MRI markers of clinically established MSA | 3. Hypometabolism on FDG-PET in the putamen* | ||||
| For MSA-P | 4. Presynaptic nigrostriatal dopaminergic denervation on SPECT or PET* | ||||
| 1. Atrophy of: | Supportive features | ||||
| ● Putamen (and a signal decrease on iron-sensitive sequences) | 1. Orofacial dystonia | ||||
| ● Middle cerebellar peduncle | 2. Disproportionate antecollis | ||||
| ● Pons | 3. Camptocormia and/or Pisa syndrome | ||||
| ● Cerebellum | 4. Contractures of hands or feet | ||||
| 2. “Hot cross bun” sign | 5. Inspiratory sighs | ||||
| 3. Increased diffusivity of: | 6. Severe dysphonia | ||||
| ● Putamen | 7. Severe dysarthria | ||||
| ● Middle cerebellar peduncle | 8. New or increased snoring* | ||||
| For MSA-C | 9. Cold hands and feet | ||||
| 1. Atrophy of: | 10. Pathologic laughter or crying | ||||
| ● Putamen (and a signal decrease on iron-sensitive sequences) | 11. Jerky, myoclonic postural/action tremor | ||||
| ● Infratentorial structures (pons and middle cerebellar peduncle) | Nonsupporting features | ||||
| “Hot cross bun” sign | 1. Classic pill-rolling rest tremor* | ||||
| 2. Increased diffusivity of: | 2. Clinically significant neuropathy* | ||||
| ● Putamen | 3. Hallucinations not induced by drugs | ||||
| Exclusion criteria | 4. Onset after age 75 yr* | ||||
| 1. Substantial and persistent beneficial response to dopaminergic medications | 5. Family history of ataxia or parkinonsinsm | ||||
| 2. Unexplained anosmia on olfactory testing | 6. Dementia (on DSM-IV) | ||||
| 3. Fluctuating cognition with pronounced variation in attention and alertness and early decline in visuoperceptual abilities | 7. White matter lesions suggesting multiple sclerossis | ||||
| 4. Recurrent visual hallucinations not induced by drugs within 3 years of disease onset | |||||
| 5. Dementia according to DSM-V within 3 years of disease onset | |||||
| 6. Downgaze supranuclear palsy or slowing of vertical saccades | |||||
| 7. Brain MRI findings suggestive of an alternative diagnosis (e.g., PSP, multiple sclerosis, vascular parkinsonism, symptomatic cerebellar disease, etc.) | |||||
| 8. Documentation of an alternative condition (conditions mimicking MSA, including genetic or symptomatic ataxia and parkinsonism) known to produce autonomic failure, ataxia, or parkinsonism and plausibly connected to the patient’s symptoms | |||||
* not included in the new diagnostic criteria.
MDS, Movement Disorder Society; MSA, multiple system atrophy; MSA-P, MSA-parkinsonian type; MSAC, MSA-cerebellar type; DSM, Diagnostic & Statistical Manual of Mental Disorders; PSP, progressive supranuclear palsy; FDG, [18F]Fluorodeoxyglucose; PET, positron emission tomography; SPECT, single photon emission computed tomography.
- 1. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 2. Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020;585:464–469.ArticlePubMedPMCPDF
- 3. Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC, et al. α-synuclein strains target distinct brain regions and cell types. Nat Neurosci 2020;23:21–31.ArticlePubMedPDF
- 4. Williams GP, Marmion DJ, Schonhoff AM, Jurkuvenaite A, Won WJ, Standaert DG, et al. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol 2020;139:855–874.ArticlePubMedPMCPDF
- 5. Jucaite A, Cselényi Z, Kreisl WC, Rabiner EA, Varrone A, Carson RE, et al. Glia imaging differentiates multiple system atrophy from Parkinson’s disease: a positron emission tomography study with [11C]PBR28 and machine learning analysis. Mov Disord 2022;37:119–129.PubMed
- 6. Shadrin AA, Mucha S, Ellinghaus D, Makarious MB, Blauwendraat C, Sreelatha AAK, et al. Shared genetics of multiple system atrophy and inflammatory bowel disease. Mov Disord 2021;36:449–459.ArticlePubMedPDF
- 7. Ferreira N, Gram H, Sorrentino ZA, Gregersen E, Schmidt SI, Reimer L, et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol 2021;142:87–115.ArticlePubMedPMCPDF
- 8. Pérez-Soriano A, Bravo P, Soto M, Infante J, Fernández M, Valldeoriola F, et al. MicroRNA deregulation in blood serum identifies multiple system atrophy altered pathways. Mov Disord 2020;35:1873–1879.ArticlePubMedPDF
- 9. Meissner WG, Traon AP, Foubert-Samier A, Galabova G, Galitzky M, Kutzelnigg A, et al. A phase 1 randomized trial of specific active α-synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord 2020;35:1957–1965.ArticlePubMedPMCPDF
- 10. Shin JY, Lee PH. Mesenchymal stem cells modulate misfolded α-synuclein in parkinsonian disorders: a multitarget disease-modifying strategy. Stem Cell Res 2020;47:101908.ArticlePubMed
- 11. Singer W, Dietz AB, Zeller AD, Gehrking TL, Schmelzer JD, Schmeichel AM, et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology 2019;93:e77–e87.ArticlePubMedPMC
- 12. Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol 2012;72:32–40.ArticlePubMed
- 13. Rascol O, Cochen de Cock V, Pavy-Le Traon A, Foubert-Samier A, Thalamas C, Sommet A, et al. Fluoxetine for the symptomatic treatment of multiple system atrophy: the MSA-FLUO trial. Mov Disord 2021;36:1704–1711.ArticlePubMedPDF
- 14. McCarter SJ, Coon EA, Savica R, St Louis EK, Bower JH, Benarroch EE, et al. Lower vitamin B12 level at multiple system atrophy diagnosis is associated with shorter survival. Mov Disord 2020;35:1462–1466.ArticlePubMedPMCPDF
- 15. Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord 2022;37:1131–1148.ArticlePubMedPMCPDF
- 16. Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020;578:273–277.ArticlePubMedPMCPDF
- 17. Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orrù CD, et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 2020;140:49–62.ArticlePubMedPMCPDF
- 18. Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A, Querejeta-Coma A, et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 2022;145:584–595.ArticlePubMedPDF
- 19. Hall S, Orrù CD, Serrano GE, Galasko D, Hughson AG, Groveman BR, et al. Performance of αSynuclein RT-QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun 2022;10:90.ArticlePubMedPMCPDF
- 20. Donadio V, Incensi A, Rizzo G, De Micco R, Tessitore A, Devigili G, et al. Skin biopsy may help to distinguish multiple system atrophy–parkinsonism from Parkinson’s disease with orthostatic hypotension. Mov Disord 2020;35:1649–1657.ArticlePubMedPDF
- 21. Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Boeve BF, Sletten DM, et al. Alpha‐synuclein oligomers and neurofilament light chain in spinal fluid differentiate multiple system atrophy from Lewy body synucleinopathies. Ann Neurol 2020;88:503–512.ArticlePubMedPMCPDF
- 22. Zhang L, Cao B, Hou Y, Gu X, Wei Q, Ou R, et al. Neurofilament light chain predicts disease severity and progression in multiple system atrophy. Mov Disord 2022;37:421–426.ArticlePubMedPDF
- 23. Krismer F, Beliveau V, Seppi K, Mueller C, Goebel G, Gizewski ER, et al. Automated analysis of diffusion‐weighted magnetic resonance imaging for the differential diagnosis of multiple system atrophy from Parkinson’s disease. Mov Disord 2021;36:241–245.ArticlePubMedPDF
- 24. Archer DB, Bricker JT, Chu WT, Burciu RG, McCracken JL, Lai S, et al. Development and validation of the automated imaging differentiation in parkinsonism (AID-P): a multicentre machine learning study. Lancet Digit Health 2019;1:e222–e231.ArticlePMC
- 25. Ebina J, Hara K, Watanabe H, Kawabata K, Yamashita F, Kawaguchi A, et al. Individual voxel-based morphometry adjusting covariates in multiple system atrophy. Parkinsonism Relat Disord 2021;90:114–119.ArticlePubMed
- 26. Gandor F, Vogel A, Claus I, Ahring S, Gruber D, Heinze HJ, et al. Laryngeal movement disorders in multiple system atrophy: a diagnostic biomarker? Mov Disord 2020;35:2174–2183.ArticlePubMedPMCPDF
- 27. Lee YH, Ando T, Lee JJ, Baek MS, Lyoo CH, Kim SJ, et al. Later-onset multiple system atrophy: a multicenter Asian study. Mov Disord 2020;35:1692–1693.ArticlePubMedPDF
- 28. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083.PubMed
- 29. Watanabe H, Riku Y, Hara K, Kawabata K, Nakamura T, Ito M, et al. Clinical and imaging features of multiple system atrophy: challenges for an early and clinically definitive diagnosis. J Mov Disord 2018;11:107–120.ArticlePubMedPMCPDF
- 30. Schrag A, Bohlken J, Kostev K. Pre-diagnostic presentations of multiple system atrophy case control study in a primary care dataset. Parkinsonism Relat Disord 2022;99:101–104.ArticlePubMed
- 31. Osaki Y, Morita Y, Miyamoto Y, Ohtsuru S, Shogase T, Furushima T, et al. Identification of a pre-possible multiple system atrophy phase. Acta Neurol Scand 2021;143:313–317.ArticlePubMedPDF
- 32. Eschlböck S, Delazer M, Krismer F, Bodner T, Fanciulli A, Heim B, et al. Cognition in multiple system atrophy: a single-center cohort study. Ann Clin Transl Neurol 2020;7:219–228.ArticlePubMedPMCPDF
- 33. Miki Y, Foti SC, Hansen D, Strand KM, Asi YT, Tsushima E, et al. Hippocampal α-synuclein pathology correlates with memory impairment in multiple system atrophy. Brain 2020;143:1798–1810.ArticlePubMedPDF
- 34. Teil M, Dovero S, Bourdenx M, Arotcarena ML, Camus S, Porras G, et al. Brain injections of glial cytoplasmic inclusions induce a multiple system atrophy-like pathology. Brain 2022;145:1001–1017.ArticlePubMedPDF
- 35. Mészáros L, Hoffmann A, Wihan J, Winkler J. Current symptomatic and disease-modifying treatments in multiple system atrophy. Int J Mol Sci 2020;21:2775.ArticlePubMedPMC
- 36. Levin J, Maaß S, Schuberth M, Giese A, Oertel WH, Poewe W, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2019;18:724–735.ArticlePubMed
- 37. Palma JA, Martinez J, Millar Vernetti P, Ma T, Perez MA, Zhong J, et al. mTOR inhibition with sirolimus in multiple system atrophy: a randomized, double-blind, placebo-controlled futility trial and 1-year biomarker longitudinal analysis. Mov Disord 2022;37:778–789.PubMedPMC
- 38. Grimaldi S, Boucekine M, Witjas T, Fluchere F, Azulay JP, Guedj E, et al. Early atypical signs and insula hypometabolism predict survival in multiple system atrophy. J Neurol Neurosurg Psychiatry 2021;92:881–889.Article
- 39. Meyer M, Lamare F, Asselineau J, Foubert-Samier A, Mazère J, ZanottiFregonara P, et al. Brain 5-HT1A receptor binding in multiple system atrophy: an [18F]-MPPF PET study. Mov Disord 2021;36:246–251.ArticlePubMedPDF
- 40. Riku Y, Watanabe H, Mimuro M, Iwasaki Y, Ito M, Katsuno M, et al. Non-motor multiple system atrophy associated with sudden death: pathological observations of autonomic nuclei. J Neurol 2017;264:2249–2257.ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
- A Blinded Evaluation of Brain Morphometry for Differential Diagnosis of Atypical Parkinsonism
Kazuya Kawabata, Florian Krismer, Beatrice Heim, Anna Hussl, Christoph Mueller, Christoph Scherfler, Elke R. Gizewski, Klaus Seppi, Werner Poewe
Movement Disorders Clinical Practice.2024; 11(4): 381. CrossRef - The potential of phosphorylated α‐synuclein as a biomarker for the diagnosis and monitoring of multiple system atrophy
Toufik Abdul‐Rahman, Ranferi Eduardo Herrera‐Calderón, Arjun Ahluwalia, Andrew Awuah Wireko, Tomas Ferreira, Joecelyn Kirani Tan, Maximillian Wolfson, Shankhaneel Ghosh, Viktoriia Horbas, Vandana Garg, Asma Perveen, Marios Papadakis, Ghulam Md Ashraf, Ath
CNS Neuroscience & Therapeutics.2024;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite