E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 6(1); 2013 > Article
-
Review Article
New Perspective on Parkinsonism in Frontotemporal Lobar Degeneration - Hee Kyung Parka, Sun J. Chungb
-
Journal of Movement Disorders 2013;6(1):1-8.
DOI: https://doi.org/10.14802/jmd.13001
Published online: April 30, 2013
aDepartment of Neurology, Inje University Ilsan Paik Hospital, Goyang, Korea
bDepartment of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding author: Sun J. Chung, MD, PhD Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 138-736, Korea, Tel +82-2-3010-3440, Fax +82-2-474-4691, E-mail sjchung@amc.seoul.kr
- The authors have no financial conflicts of interest.
• Received: February 18, 2013 • Accepted: March 1, 2013
Copyright © 2013 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 24,327 Views
- 235 Download
- 25 Crossref
ABSTRACT
- Frontotemporal dementia (FTD) is the second most common type of presenile dementia. Three clinical prototypes have been defined; behavioral variant FTD, semantic dementia, and progressive nonfluent aphasia. Progressive supranuclear palsy, corticobasal degeneration, and motor neuron disease may possess clinical and pathological characteristics that overlap with FTD, and it is possible that they may all belong to the same clinicopathological spectrum. Frontotemporal lobar degeneration (FTLD) is a clinicopathological syndrome that encompasses a heterogenous group of neurodegenerative disorders. Owing to the advancement in the field of molecular genetics, diagnostic imaging, and pathology, FTLD has been the focus of great interest. Nevertheless, parkinsonism in FTLD has received relatively less attention. Parkinsonism is found in approximately 20–30% of patients in FTLD. Furthermore, parkinsonism can be seen in all FTLD subtypes, and some patients with familial and sporadic FTLD can present with prominent parkinsonism. Therefore, there is a need to understand parkinsonism in FTLD in order to obtain a better understanding of the disease. With regard to the clinical characteristics, the akinetic rigid type of parkinsonism has predominantly been described. Parkinsonism is frequently observed in familial FTD, more specifically, in FTD with parkinsonism linked to chromosome 17q (FTDP-17). The genes associated with parkinsonism are microtubule associated protein tau (MAPT), progranulin (GRN or PGRN), and chromosome 9 open reading frame 72 (C9ORF72) repeat expansion. The neural substrate of parkinsonism remains to be unveiled. Dopamine transporter (DAT) imaging revealed decreased uptake of DAT, and imaging findings indicated atrophic changes of the basal ganglia. Parkinsonism can be an important feature in FTLD and, therefore, increased attention is needed on the subject.
- Since Neary et al.3 proposed the clinical diagnostic criteria of three clinical syndromes in FTLD many researchers have used those criteria in order to clarify the syndromes.14 This classification system of FTD consists of behavioral and language variants of FTD. The language variants of FTD include PNFA and SD. Continuous research in the field of genetics and pathology related to FTD has disclosed that other clinical entities such as MND or PSP or CBD may be the presenting symptom of FTD. While the afore-mentioned categorization may suffice from the standpoint of cognitive neuroscience, a movement specialist may demand more understanding of motor aspect. In this regard, we would like to add the motor variant of FTD. PSP or CBD may share common clinical features and pathological findings with FTD.5 On the basis of the criteria proposed by Neary et al.,3 physical signs in FTD include parkinsonism, comprising akinesia, rigidity, and tremor. Additionally, patients with MND may develop FTD during the course of their diseases, while patients with FTD may present with symptoms of MND.15,16 FTD and MND may share the pathological features including the presence of TAR DNA-binding protein (TDP-43)-immunoreactive inclusions.17 An updated classification that includes the motor variant of FTD, which is further divided into parkinsonism-predominant and MND-predominant groups, may offer further insight into the clinical presentations of FTD (Fig. 1).
Clinical Classification of FTLD
- There have been few studies focusing on the prevalence of parkinsonism in sporadic FTD (Table 1).18 The researchers paid little attention to parkinsonism in FTLD and, therefore the exact prevalence and incidence of parkinsonism in FTLD has not been revealed. In a study that was published in 2004, thirty percent (n=18) of patients with pathologically proven FTLD showed extrapyramidal signs, in particular, rigidity or akinesia.4 Five patients with bvFTD, one patient with PNFA, three patients with FTD-MND, and nine patients with CBD initially presented with parkinsonism. In this report, none of the patients with SD demonstrated parkinsonism as the presenting symptom.4
- Piguet et al.14 showed that less than 10% of patients with bvFTD are likely to demonstrate parkinsonism at the beginning. Even after disease progression, less than 20% of these patients eventually show parkinsonism. The results which was reported by Rascovsky et al.19 was similar. In one big cohort consisting of 364 patients with FTD, sixteen percent (n=57) displayed parkinsonism.20 In this report, from the clinical standpoint, early parkinsonism was seen in 18% (n=45) of bvFTD patients, 11% (n=6) of SD patients, and 14% (n= 5) of PNFA patients, while it was shown in only 4% (n=1) of FTD-MND patients.20 Meanwhile, genetically, parkinsonism was reported in seven patients with MAPT mutations, five patients with PGRN mutations, and five patients with an autosomal dominant form of an unknown genetic defect.20 However, this study did not show a difference in the frequency of parkinsonism of the four clinical subtypes including bvFTD, SD, PNFA, and FTD-MND between sporadic FTD and familial FTD. In a study of 56 FTD-MND patients parkinsonism was seen in one patient with behavioral-dominant FTD-MND and six patients with language-dominant FTD-MND.21 In this case series, they found that psychosis was more common in behavioral-dominant FTD-MND while parkinsonism and limb apraxia were more common in language-dominant FTDMND.21 Padovani et al.18 focused on extrapyramidal symptoms in 75 patients with bvFTD, among whom 22.7% (n=17) showed early parkinsonism. Another study evaluating parkinsonism in FTD revealed that 22% (70/319) of patients demonstrated parkinsonism. The authors divided FTD patients with parkinsonism into two groups: those who initially presented with the movement disorder (15 CBDs and 7 PSPs) and those who initially presented with a cognitive disorder followed by the development of parkinsonism (n=48).13 Regarding the presenting symptoms, Padovani et al.18 reported that parkinsonism occurred as a presenting symptom in 22.7% of their patients, while Kertesz et al.13 observed that parkinsonism as a presenting symptom in only 6% (n=22) of their patients.
- As far as we know, there has been no study has reporting the survival analysis between FTD patients with and without parkinsonism. There is still need for a large, prospective, multi-center cohort study focusing on the prevalence of parkinsonism in FTD. In addition, there has been a lack of consistency due to various methodological issues, deriving from different regions of interest between movement specialists and researchers specializing in cognitive neurology, ambiguity in the definition of parkinsonism, and diversity in patient characteristics including disease duration and age.
Epidemiology of Parkinsonism in FTLD
- Ever since the first report on Pick’s disease,1 there have been some case studies describing the extrapyramidal symptoms in Pick’s disease.22,23 One of these reports described a patient with a masked face, clumsiness of fingers, and cogwheel rigidity revealing extensive involvement of the caudate nucleus, the substantia nigra, the pallidum, and the subthalamic nucleus.23 The Lund and Manchester groups described parkinsonisms as late-occurring akinesia, rigidity, and tremor in 1994.2 Four years later, Neary et al.3 stated that parkinsonian signs (akinesia, rigidity, tremor) typically emerge only during late disease. Representative clinical subtypes in FTD with parkinsonism consist of PSP, CBD, and FTDP-17. Clinical and pathologic manifestations in CBD, PSP, and FTD are diverse.
- Progressive supranuclear palsy may have different pathologic findings from the clinical phenotypes.24 The phenotypes of PSP may be heterogeneous.24 The classic phenotype, Richardson’s syndrome has distinctive clinical features differentiating it from Parkinson’s disease (PD) or other PSP sub-types. Vertical supranuclear gaze palsy and surprised facial appearance are characteristic features in PSP. The axial symptoms are predominant and, therefore, a lurching gait and unexpected falls can be seen. Symmetry, axial rigidity, minimal or absent tremor, and poor response to levodopa are distinguishing features of parkinsonism in PSP. Cognitive decline was observed as an early feature in 29% of patients with pathologically proven PSP and as a late feature in 74%.25 This finding is not surprising given that PSP is a clinical subtype of FTLD. Patients with pathological findings of PSP may have clinical variants of PSP such as PSP-parkinsonism (PSP-P), PSP-pure akinesia with gait freezing (PSP-PAGF), PSP-corticobasal syndrome (PSP-CBS), and PSP-progressive non-fluent aphasia (PSP-PNFA).24
- Corticobasal degeneration differs from PD or PSP in terms of clinical manifestations. The clinical diagnostic criteria includes a strong degree of asymmetry with rigidity, bradykinesia, atypical tremor (postural and action), alien limb phenomenon, dystonia, myoclonus, and cortical signs (myoclonus, apraxia).26 However, there have been a lot of discrepant results between clinical and pathological diagnoses.27–29 Josephs et al.5 reported that among cases that were clinically diagnosed as CBD half were pathologically proven to be CBD, while the remaining half were pathologically proven to be PSP. By contrast, 90% of cases that were clinically diagnosed to be PSP were pathologically proven to be PSP.5 In a recent study comprising 21 patients clinically diagnosed with CBS, only five patients were pathologically diagnosed to have CBD.28 The other pathological diagnoses were PSP, Alzheimer’s disease (AD), PD, and FTD. Inversely, 42% of CBD patients had been diagnosed with PSP. The overall sensitivity in predicting CBD pathology was 47%.28 In this study, the authors classified CBD into CBD-CBS and CBD-Richardson’s syndrome according to the clinical features.28
- FTD with parkinsonism linked to chromosome 17 with MAPT mutation develops at an average age of 49 years and presents with behavioral change, dementia, and parkinsonism.6,12,30 It is not possible to correlate the clinical manifestations with the genetic subtypes. However, it has been previously shown that the dementia-dominant group correlated with the H1/H2 genotype, while the parkinsonism-dominant group correlated with the H1/H1 genotype.31,32 The P301L and exon 10+6 mutation carriers often present with behavioral or personality change.33 The N279K mutation carriers present with parkinsonism, which consists of rigidity, bradykinesia, falls, vertical gaze palsy, and poor response to levodopa.34 FTDP-17 with PGRN mutation develops at a mean age of 59 years and presents with various symptoms. Behavioral or personality changes and language impairment may be seen as the initial manifestation.6,30 As the disease progresses, markedly asymmetric parkinsonism such as bradykinesia or rigidity, which is similar to that observed in CBD develops.35,36 In 2011, an important mutation, an expansion of a non-coding GGGGCC hexanucleotide repeat in the C9ORF72 gene, was identified in familial FTD and ALS.37,38 Twelve of 30 patients with the C9ORF72 mutation showed parkinsonism during the disease course, and in addition, one patient presented with parkinsonism.39 In this report, parkinsonism in eight of twelve patients was predominantly akinetic rigidity, while remaining four patients demonstrated resting tremor.39
- Initial manifestations of bvFTD are apathy, disinhibition, repetitive or stereotypic behavior, lack of mental flexibility, changes in eating behavior, decline in personal hygiene, and loss of sympathy.3,40 Physical signs include primitive reflexes in 40% of patients and parkinsonism in less than 10%.14 Parkinsonism in patients with PNFA may present with PSP-PNFA or CBS-PNFA and is typically akinetic-rigid type of parkinsonism.5,41 One study reported that the nonfluent/agrammatic variant of primary progressive aphasia (PPA) had significantly more parkinsonian motor features than the logopenic PPA variant.42 However, when their parkinsonian motor features were analyzed, there was no difference in gait/posture and tremor subscales between the two variants.42 SD with parkinsonism is not yet fully understood. One study reported that six 56 SD patients showed parkinsonism, but the details of their clinical features were not described.20 In the case of FTD-MND, about 12% of the patients demonstrated parkinsonism in one report, but no details were presented.21
Clinical Characteristics
- Family history is found in approximately 45% of patients with FTD43 and autosomal dominant inheritance is found in 10%.44 Familial FTLD is more common in bvFTD, and is less common in SD and FTD-MND.44 Familial forms of FTD associated with parkinsonism are caused by two common mutations (MAPT and PGRN) and one less common genetic mutation (CHMP2B). As mentioned earlier, carriers of the C9ORF72 gene mutaion may be associated with parkinsonism.39 Severeal disorders associated with FTDP include familial PSP, hereditary diffuse leukoencephalopathy with axoal spheroids (HDLS), and neurodegenerative overlap syndrome.45
- Familial FTDP associated with chromosome 17 is caused by MAPT mutations and PGRN mutations. Fourty-four pathological mutations (http://www.molgen.ua.ac.be/FTDMutations) and two extended haplotypes, H1 and H2, have been identified in the MAPT gene. H1 haplotype is hyperexpressed in CBD and PSP, which indicates its susceptibility to 4R tauopathy.46 An association has been found between the H1/H1 genotype and the parkinsonism-plus-predominant phenotype in carriers of MAPT mutations.31,32 The discovery of PGRN mutations has been made in 2006.47 Since then, 67 pathological mutations (http://www.molgen.ua.ac.be/FTDMutations) have been found. PGRN mutation carriers may demonstrate various clinical manifestations, although the exact correlation between the genotype and phenotype cannot be made.48 One fourth of the patients with PGRN mutation may show manifestations similar to patients with PNFA; however, progressive aphasia in these patients usually does not include apraxia of speech.49
- The FUS gene mutation was first reported in familial and sporadic ALS in 2009.50–52 However, it has rarely been observed in patients with bvFTD53 or FTLD-ALS.54–56 One patient with FUS mutation developed ALS with gait and speech difficulty, while the patient’s brother was diagnosed with parkinsonism and dementia.54 Reports on parkinsonism in carriers of the FUS mutation have been limited. Expanded GGGGCC repeat in C9ORF72 is an important cause of FTD and ALS.37,38 The identification of C9ORF72 repeat expansions adds FTD-MND to the category of noncoding repeat expansion disorders, for example, spinocerebellar ataxia (SCA8, SCA31, SCA36),57–59 myotonic dystrophies (DM1 and DM2),60,61 and fragile-X associated tremor/ataxia syndrome (FXTAS).62 Given that 48% of patients with the C9ORF72 mutation showed parkinsonism,39 further research may identify this gene as an important cause of FTD with parkinsonism.
Genetic Studies
- Pathological findings in FTLD can be subclassified according to the accumulated protein: FTLD with tau inclusions (FTLD-tau), FTLD with tau-negative and TDP-43-positive inclusions (FTLD-TDP), and FTLD with tau/TDP-43 negative and FUS-positive inclusions (FTLD-FUS).63 Based on five large clinicopathologic studies,4,5,64–66 the most common pathology in FTLD-tau was CBD (35%), followed by PSP (31%), Pick’s disease (30%), and agyrophilic grain disease (4%).67 FTLD-TDP consists of four subtype; FTLD-TDP type 1, 2, 3, 4.63 FTLD-tau pathology are mostly associated with clinical syndromes, bvFTD, PNFA, PSP, CBD, and FTLD-17 (MA-PT), while FTLD-TDP pathology are related with bvFTD, SD, CBS, FTDP-17 (PGRN), and FTD-MND.67 FTLD-tau pathology is rarely found in patients with SD while FTLDTDP pathology is uncommon in patients with PNFA.67 It seems that prominent parkinsonism is most likely associated with FTLD-tau pathology.64,67 FTLD-FUS pathology may be found in patients with bvFTD.68 The burden of FUS pathology was found to be moderate not only in the frontal and temporal neocortex but also in the striatum.68 Patients with the C9ORF72 mutation did not show correlation between the degree of extrapyramidal dysfunction and any measure of pathology in the striatum or substantia nigra.39 One patient with the C9OR72 expansion who was diagnosed with both PD and ALS had neuropathological features of both PD and ALS including cell loss from the substantia nigra and 6/6 Braak grade α-synuclein pathology.69
Pathology
- The brain magnetic resonance imaging (MRI) performed in patients with FTD typically reveals prominent asymmetric atrophy of the frontal and temporal lobes.70 Fluorodeoxyglucose positron emission tomography (FDG PET) can improve diagnostic accuracy by revealing hypometabolism in those areas.71 Various findings have been reported in the clinical subtypes of FTD (Fig. 2). Progressive degeneration in bvFTD can be found in anterior cingulate cortex (ACC), frontal insular (FI), rostromedial prefrontal cortex (PFC), frontal pole (FP), and ventral striatum.70,72 Salience network connectivity in bvFTD patients was dramatically attenuated in the frontoinsular area and temporal pole.73 Furthermore, bvFTD patients demonstrated striking disruption of salience network in the brainstem, limbic, and subcortical structures including substantia nigra, ventral tegmental area, nucleus accumbens, ventral striatopallidum, and thalamus.73 SD patients demonstrate remarkably asymmetric atrophic changes in ventromedial PFC, ACC, FI, and ventral striatum.74,75 With increasing disease severity, atrophic changes on the contralateral side become noticeable.75 The involvement in the frontal operculum, supplementary motor area, and dorsal insula are observed in patients with PNFA.76,77
- Patients with bvFTD and FUS pathology demonstrate a distinct pattern of atrophy, with severe caudate atrophy, compared to the patients with FTLD-tau or FTLD-TDP pathology.78 When it comes to the comparison between patients with PGRN mutations and MAPT mutations, patients with PGRN mutations demonstrate more asymmetric atrophy in the frontal, temporal, and inferior parietal lobes, while patients with MAPT mutations show relatively symmetric atrophic changes in the anteromedial temporal area and orbitofrontal cortex.79,80 Although previous imaging study reported the involvement of substantia nigra,73 there has been no study focusing the correlation between parkinsonism and structural lesion in FTD.
- Amyloid imaging studies can differentiate FTD from AD and dementia with Lewy bodies (DLB).81,82 In a study by Rowe et al.81 comprising 6 patients with FTD, no retention of Pittsburgh compound B (PIB) was seen. Furthermore, Engler et al.82 reported that eight of ten patients with FTD showed little retention of PIB. Therefore, amyloid PET in FTD patients with parkinsonism may potentially help differentiate FTD from DLB or vice versa (Fig. 3). However, we should consider that amyloid PET findings in DLB may be variable. Dopamine transporter imaging studies in the past revealed decreased uptake in bilateral putamina, and the correlation between the uptake ratio and parkinsonian motor status has been shown.83,84
Neuroimaging Studies
- Parkinsonism in FTLD may be an important clinical characteristic not only as a presenting feature of the PSP and CBD, but also as an accompanying feature in other subtypes of FTLD. Thus far, we do not know the exact anatomic substrate nor do we understand the pathomechanism of parkinsonism in FTLD. In the future, further researches focusing on parkinsonism in FTLD should be carried out in order to obtain a holistic understanding of FTLD.
Conclusion
- This study was supported by a grant of the Korea Health 21 R&D Project, Ministry of Health, Welfare, and Family Affairs, Republic of Korea (A102065).
Acknowledgments
Figure 1.Classification of frontotemporal lobar degeneration based on the clinical manifestations. FTLD: frontotemporal lobar denetation, bvFTD: behavioral variant frontotemporal dementia, PNFA: progressive nonfluent aphasia, SD: semantic dementia, PSP: progressive supranuclear palsy, CBD: corticobasal degeneration, FTDP-17: frontotemporal dementia with parkinsonism linked to chromosome 17, FTD-MND: frontotemporal dementia-motor neuron disease.
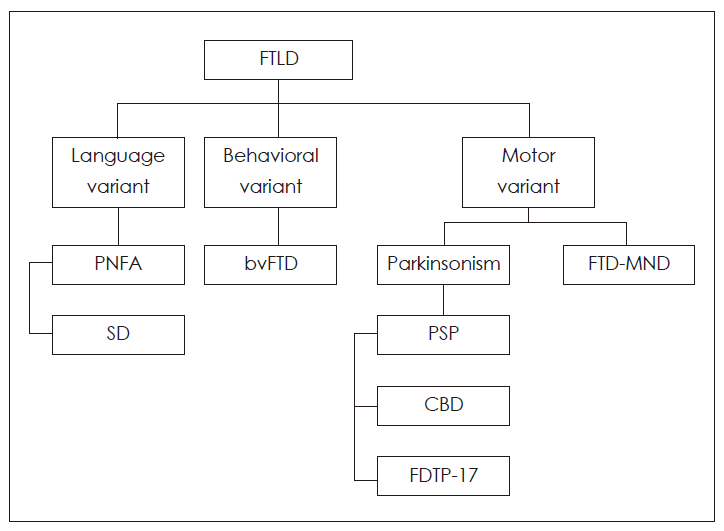
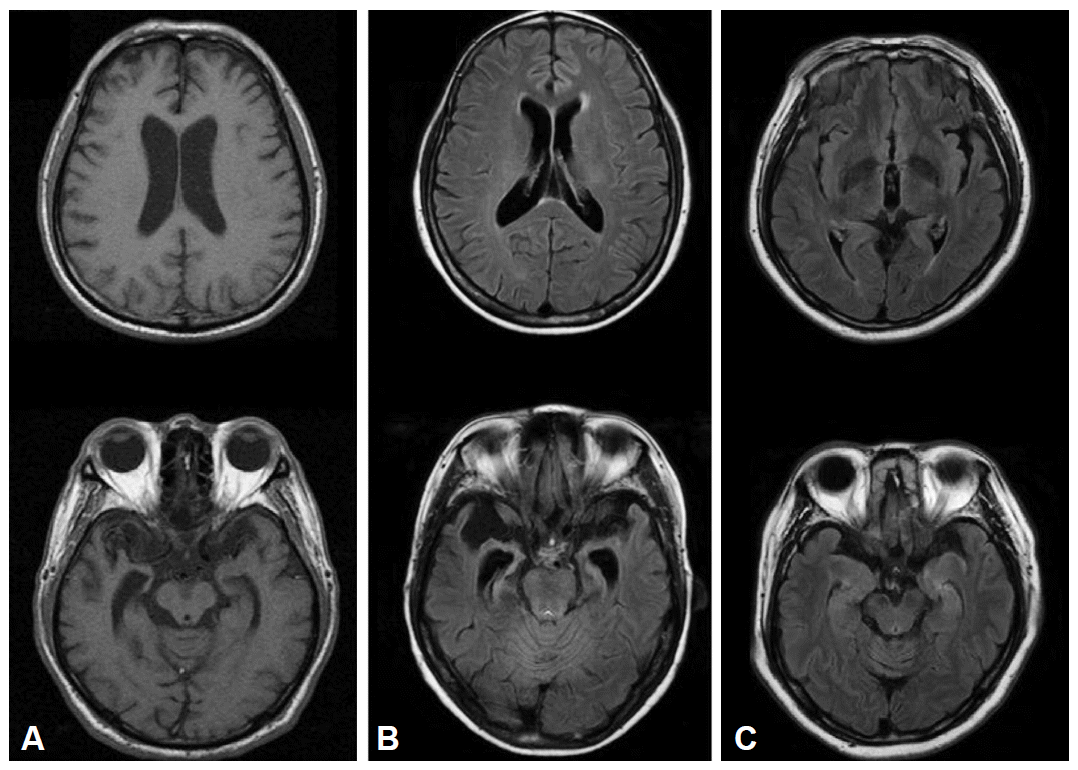
Figure 2.Brain MRI in frontotemporal dementia patients. A: A patient with behavioral variant frontotemporal dementia who presented with parkinsonism showed frontal and temporal atrophy. B: The presence of symmetric atrophy in bilateral temporal lobes was seen in a semantic dementia case. C: A patient with progressive nonfluent aphasia demonstrated the focal atrophy of the left perisylvian area.
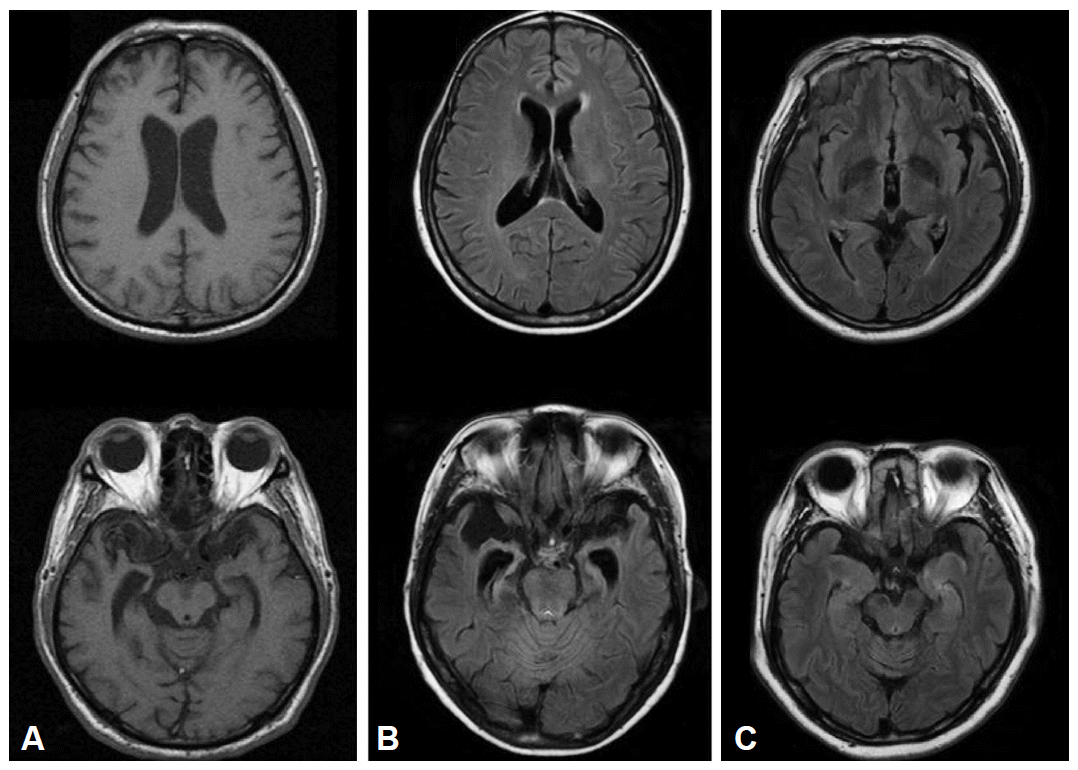
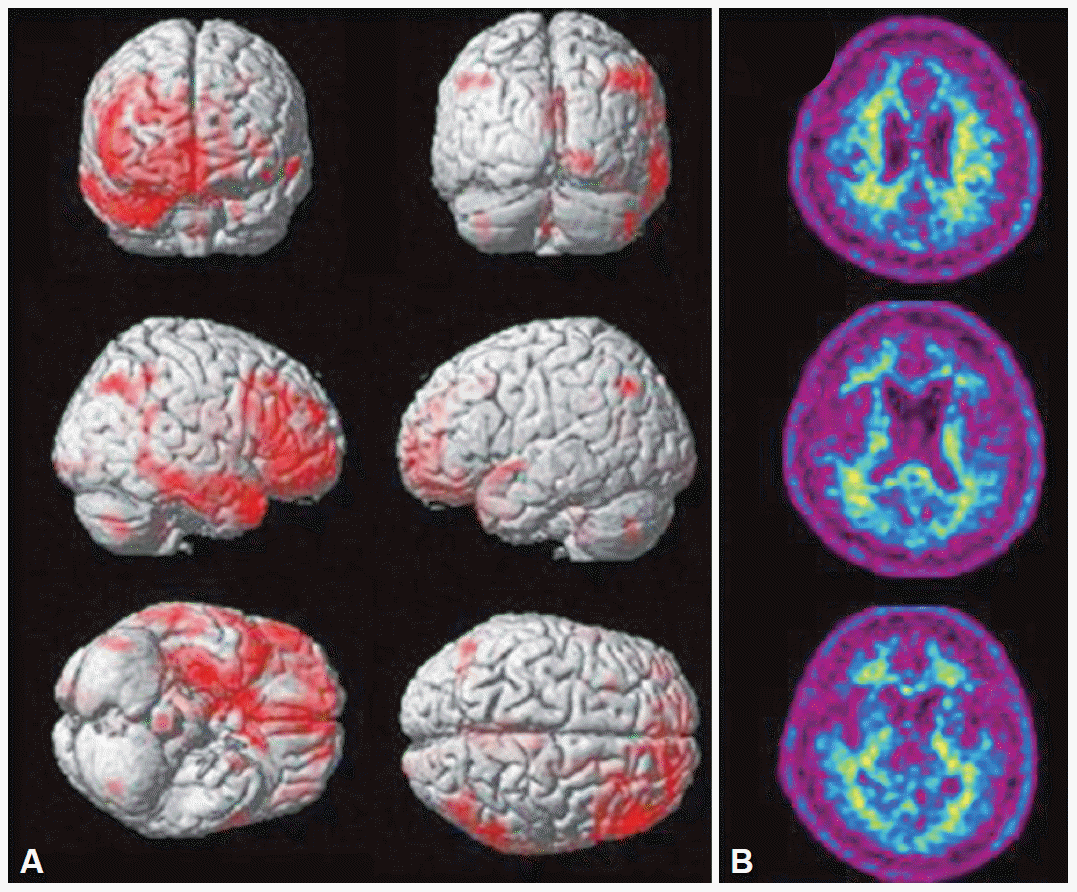
Figure 3.Positron emission tomography (PET) images in a frontotemporal dementia (FTD) patient. This FTD patient who presented with parkinsonism demonstrated frontotemporal hypometabolism in fluorodeoxyglucose positron emission tomography (FDG PET) (A). [11C] Pittsburgh compound-B (PIB) PET showed little PIB retention.

Table 1.Clinical characteristics of patients with frontotemporal dementia who demonstrated parkinsonism
| Frequency | Parkinsonism | Clinical classification | Pathology | |
|---|---|---|---|---|
| Hodges et al.4 | 30% (n=18/61) | Rigidity or akinesia | bvFTD (5), SD (0), PNFA (1), FTD-MND (3), CBD(9) | Yes |
| Piguet et al.14 | <20% | Lack of detailed description | bvFTD | Yes: 18/45 |
| Rascovsky et al.19 | <20% | Akinesia, rigidity, tremor | bvFTD | Yes: 176/176 |
| Seelaar et al.20 | 16% (n=57/364) | Two of four clinical signs (rigidity, tremor, bradykinesia, and postural instability) | bvFTD (45), SD (6), PNFA (5), FTD-MND (1) | Yes: 35/364 |
| Coon et al.21 | 12.5% (n=7/56) | Lack of detailed description | FTD-MND (7) | No |
| Padovani et al.18 | 22.7% (n=17/75) | Based on UPDRS rating | bvFTD (17) | No |
| Kertesz et al.13 | 22% (n=70/319) | Lack of detailed description | CBD (15), PSP (7), aphasic, behavioral or both (48) | No |
- 1. Pick A. Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prag Med Wochenschr 1892;165–167.
- 2. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 1994;57:416–418.ArticlePubMedPMC
- 3. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554.ArticlePubMed
- 4. Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406.ArticlePubMed
- 5. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48.ArticlePubMed
- 6. Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch Neurol 2008;65:460–464.ArticlePubMedPMC
- 7. Seltman RE, Matthews BR. Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs 2012;26:841–870.ArticlePubMed
- 8. Tartaglia MC. Frontotemporal lobar degeneration: new understanding brings new approaches. Neuroimaging Clin N Am 2012;22:83–97.viii.ArticlePubMed
- 9. Arvanitakis Z. Update on frontotemporal dementia. Neurologist 2010;16:16–22.ArticlePubMedPMC
- 10. Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 2011;82:476–486.ArticlePubMed
- 11. Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010;24:375–398.ArticlePubMedPMC
- 12. Espay AJ, Litvan I. Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci 2011;45:343–349.ArticlePubMedPMC
- 13. Kertesz A, McMonagle P, Jesso S. Extrapyramidal syndromes in frontotemporal degeneration. J Mol Neurosci 2011;45:336–342.ArticlePubMed
- 14. Piguet O, Hornberger M, Shelley BP, Kipps CM, Hodges JR. Sensitivity of current criteria for the diagnosis of behavioral variant frontotemporal dementia. Neurology 2009;72:732–737.ArticlePubMedPMC
- 15. Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol 2007;64:530–534.ArticlePubMed
- 16. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 2007;6:994–1003.ArticlePubMed
- 17. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133.ArticlePubMed
- 18. Padovani A, Agosti C, Premi E, Bellelli G, Borroni B. Extrapyramidal symptoms in Frontotemporal Dementia: prevalence and clinical correlations. Neurosci Lett 2007;422:39–42.ArticlePubMed
- 19. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(Pt 9):2456–2477.ArticlePubMedPMC
- 20. Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, et al. Distinct genetic forms of frontotemporal dementia. Neurology 2008;71:1220–1226.ArticlePubMed
- 21. Coon EA, Sorenson EJ, Whitwell JL, Knopman DS, Josephs KA. Predicting survival in frontotemporal dementia with motor neuron disease. Neurology 2011;76:1886–1893.ArticlePubMedPMC
- 22. Löwenberg K. Pick’s disease: a clinicopathologic contribution. Arch Neurol Psychiatry 1936;36:768–789.Article
- 23. Akelaitis AJ. Atrophy of basal ganglia in Pick’s disease. A clinicopathologic study. Arch Neurol Psychiatry 1944;51:27–34.Article
- 24. Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009;8:270–279.ArticlePubMed
- 25. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005;128(Pt 6):1247–1258.ArticlePubMed
- 26. Litvan I, Agid Y, Goetz C, Jankovic J, Wenning GK, Brandel JP, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 1997;48:119–125.ArticlePubMed
- 27. Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125(Pt 4):861–870.ArticlePubMed
- 28. Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010;133(Pt 7):2045–2057.ArticlePubMed
- 29. Murray R, Neumann M, Forman MS, Farmer J, Massimo L, Rice A, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007;68:1274–1283.ArticlePubMed
- 30. Haugarvoll K, Wszolek ZK, Hutton M. The genetics of frontotemporal dementia. Neurol Clin 2007;25:697–715.vi.ArticlePubMed
- 31. Baba Y, Tsuboi Y, Baker MC, Uitti RJ, Hutton ML, Dickson DW, et al. The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord 2005;11:205–208.ArticlePubMed
- 32. Baba Y, Baker MC, Le Ber I, Brice A, Maeck L, Kohlhase J, et al. Clinical and genetic features of families with frontotemporal dementia and parkinsonism linked to chromosome 17 with a P301S tau mutation. J Neural Transm 2007;114:947–950.ArticlePubMed
- 33. Nasreddine ZS, Loginov M, Clark LN, Lamarche J, Miller BL, Lamontagne A, et al. From genotype to phenotype: a clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol 1999;45:704–715.ArticlePubMed
- 34. Slowinski J, Dominik J, Uitti RJ, Ahmed Z, Dickson DD, Wszolek ZK. Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 2007;27:73–80.ArticlePubMed
- 35. Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131(Pt 3):732–746.ArticlePubMed
- 36. Beck J, Rohrer JD, Campbell T, Isaacs A, Morrison KE, Goodall EF, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008;131(Pt 3):706–720.ArticlePubMedPMC
- 37. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256.ArticlePubMedPMC
- 38. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268.ArticlePubMedPMC
- 39. Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 2012;135(Pt 3):709–722.ArticlePubMedPMC
- 40. Piguet O, Hornberger M, Mioshi E, Hodges JR. Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol 2011;10:162–172.ArticlePubMed
- 41. Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol 2006;59:156–165.ArticlePubMed
- 42. Graff-Radford J, Duffy JR, Strand EA, Josephs KA. Parkinsonian motor features distinguish the agrammatic from logopenic variant of primary progressive aphasia. Parkinsonism Relat Disord 2012;18:890–892.ArticlePubMedPMC
- 43. Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol 1999;56:817–822.ArticlePubMedPMC
- 44. Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009;73:1451–1456.ArticlePubMedPMC
- 45. Fujioka S, Wszolek ZK. Clinical aspects of familial forms of frontotemporal dementia associated with parkinsonism. J Mol Neurosci 2011;45:359–365.ArticlePubMedPMC
- 46. Hutton M. Molecular genetics of chromosome 17 tauopathies. Ann N Y Acad Sci 2000;920:63–73.ArticlePubMed
- 47. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–919.ArticlePubMed
- 48. van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 2008;7:965–974.ArticlePubMed
- 49. Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 2006;129(Pt 11):3091–3102.ArticlePubMed
- 50. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208.ArticlePubMed
- 51. Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211.ArticlePubMedPMC
- 52. Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 2009;73:1176–1179.ArticlePubMedPMC
- 53. Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010;74:366–371.ArticlePubMed
- 54. Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, et al. Frame-shift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010;75:807–814.ArticlePubMedPMC
- 55. Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, et al. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry 2010;81:639–645.ArticlePubMed
- 56. Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009;73:1180–1185.ArticlePubMedPMC
- 57. Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89:121–130.ArticlePubMedPMC
- 58. Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet 2009;5:e1000600.ArticlePubMedPMC
- 59. Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 is associated with “inserted” pentanucleotide repeats containing (TGGAA)n. Am J Hum Genet 2009;85:544–557.ArticlePubMedPMC
- 60. Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992;255:1253–1255.ArticlePubMed
- 61. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864–867.ArticlePubMed
- 62. Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol 2004;1:103–105.ArticlePubMed
- 63. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119:1–4.ArticlePubMedPMC
- 64. Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962.ArticlePubMedPMC
- 65. Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005;128(Pt 9):1996–2005.ArticlePubMed
- 66. Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 2007;114:31–38.ArticlePubMed
- 67. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–153.ArticlePubMedPMC
- 68. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009;132(Pt 11):2922–2931.ArticlePubMedPMC
- 69. Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, et al. Clinicopathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012;135(Pt 3):751–764.ArticlePubMedPMC
- 70. Rabinovici GD, Seeley WW, Kim EJ, Gorno-Tempini ML, Rascovsky K, Pagliaro TA, et al. Distinct MRI atrophy patterns in autopsy-proven Alzheimer’s disease and frontotemporal lobar degeneration. Am J Alzheimers Dis Other Demen 2007;22:474–488.ArticlePubMedPMC
- 71. Foster NL, Heidebrink JL, Clark CM, Jagust WJ, Arnold SE, Barbas NR, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007;130(Pt 10):2616–2635.ArticlePubMed
- 72. Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002;58:198–208.ArticlePubMed
- 73. Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain 2010;133(Pt 5):1352–1367.ArticlePubMedPMC
- 74. Seeley WW, Bauer AM, Miller BL, Gorno-Tempini ML, Kramer JH, Weiner M, et al. The natural history of temporal variant frontotemporal dementia. Neurology 2005;64:1384–1390.ArticlePubMedPMC
- 75. Brambati SM, Rankin KP, Narvid J, Seeley WW, Dean D, Rosen HJ, et al. Atrophy progression in semantic dementia with asymmetric temporal involvement: a tensor-based morphometry study. Neurobiol Aging 2009;30:103–111.ArticlePubMedPMC
- 76. Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–346.ArticlePubMedPMC
- 77. Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129(Pt 6):1385–1398.ArticlePubMedPMC
- 78. Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack CR Jr, et al. Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol 2010;17:969–975.ArticlePubMedPMC
- 79. Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 2010;53:1070–1076.ArticlePubMedPMC
- 80. Whitwell JL, Jack CR Jr, Boeve BF, Senjem ML, Baker M, Rademakers R, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813–820.ArticlePubMedPMC
- 81. Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725.ArticlePubMed
- 82. Engler H, Santillo AF, Wang SX, Lindau M, Savitcheva I, Nordberg A, et al. In vivo amyloid imaging with PET in frontotemporal dementia. Eur J Nucl Med Mol Imaging 2008;35:100–106.ArticlePubMed
- 83. Rinne JO, Laine M, Kaasinen V, Norvasuo-Heilä MK, Någren K, Helenius H. Striatal dopamine transporter and extrapyramidal symptoms in frontotemporal dementia. Neurology 2002;58:1489–1493.ArticlePubMed
- 84. Sedaghat F, Gotzamani-Psarrakou A, Dedousi E, Arnaoutoglou M, Psarrakos K, Baloyannis I, et al. Evaluation of dopaminergic function in frontotemporal dementia using I-FP-CIT single photon emission computed tomography. Neurodegener Dis 2007;4:382–385.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Antidepressant medications in dementia: evidence and potential mechanisms of treatment-resistance
Harry Costello, Jonathan P. Roiser, Robert Howard
Psychological Medicine.2023; 53(3): 654. CrossRef - Movement disorders are linked to TDP-43 burden in the substantia nigra of FTLD-TDP brain donors
Luigi Fiondella, Priya Gami-Patel, Christian A. Blok, Annemieke J. M. Rozemuller, Jeroen J. M. Hoozemans, Yolande A. L. Pijnenburg, Marta Scarioni, Anke A. Dijkstra
Acta Neuropathologica Communications.2023;[Epub] CrossRef - Demencias degenerativas: ¿un dilema de síndromes o de enfermedades?
A. Robles Bayón
Neurología.2022; 37(6): 480. CrossRef - Degenerative dementias: a question of syndrome or disease?
A. Robles Bayón
Neurología (English Edition).2022; 37(6): 480. CrossRef - Deficient neurotransmitter systems and synaptic function in frontotemporal lobar degeneration—Insights into disease mechanisms and current therapeutic approaches
Nadine Huber, Sonja Korhonen, Dorit Hoffmann, Stina Leskelä, Hannah Rostalski, Anne M. Remes, Paavo Honkakoski, Eino Solje, Annakaisa Haapasalo
Molecular Psychiatry.2022; 27(3): 1300. CrossRef - Novel frameshift CTSF mutation causing kufs disease type B mimicking frontotemporal dementia-parkinsonism
Murat Gultekin, Zeynep Tufekcioglu, Recep Baydemir
Neurocase.2022; 28(1): 107. CrossRef - Dopamine Transporter Imaging for Frontotemporal Lobar Degeneration With Motor Neuron Disease
Ryota Kobayashi, Shinobu Kawakatsu, Makoto Ohba, Daichi Morioka, Masafumi Kanoto, Koichi Otani
Frontiers in Neuroscience.2022;[Epub] CrossRef - Investigational therapeutics for the treatment of progressive supranuclear palsy
David G Coughlin, Irene Litvan
Expert Opinion on Investigational Drugs.2022; 31(8): 813. CrossRef - Frequency and Longitudinal Course of Motor Signs in Genetic Frontotemporal Dementia
Sonja Schönecker, Francisco J. Martinez-Murcia, Boris-Stephan Rauchmann, Nicolai Franzmeier, Catharina Prix, Elisabeth Wlasich, Sandra V. Loosli, Katja Bochmann, Juan-Manuel Gorriz Saez, Robert Laforce, Simon Ducharme, Maria Carmela Tartaglia, Elizabeth F
Neurology.2022;[Epub] CrossRef - Multi-Modal Synergistic 99mTc-TRODAT-1 SPECT and MRI for Evaluation of the Efficacy of Hyperbaric Oxygen Therapy in CO-Induced Delayed Parkinsonian and Non-Parkinsonian Syndromes
Skye Hsin-Hsien Yeh, Chuang-Hsin Chiu, Hung-Wen Kao, Ching-Po Lin, Yu-Hus Lai, Wen-Sheng Huang
Antioxidants.2022; 11(11): 2289. CrossRef - Defective Lysosomal Lipid Catabolism as a Common Pathogenic Mechanism for Dementia
Jun Yup Lee, Oana C. Marian, Anthony S. Don
NeuroMolecular Medicine.2021; 23(1): 1. CrossRef - Different miRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases
Daisy Sproviero, Stella Gagliardi, Susanna Zucca, Maddalena Arigoni, Marta Giannini, Maria Garofalo, Martina Olivero, Michela Dell’Orco, Orietta Pansarasa, Stefano Bernuzzi, Micol Avenali, Matteo Cotta Ramusino, Luca Diamanti, Brigida Minafra, Giulia Peri
International Journal of Molecular Sciences.2021; 22(5): 2737. CrossRef - Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies
Katarzyna Gaweda-Walerych, Emilia Jadwiga Sitek, Ewa Narożańska, Emanuele Buratti
Cells.2021; 10(12): 3389. CrossRef - Joint contractures and acquired deforming hypertonia in older people: Which determinants?
Patrick Dehail, Nathaly Gaudreault, Haodong Zhou, Véronique Cressot, Anne Martineau, Julie Kirouac-Laplante, Guy Trudel
Annals of Physical and Rehabilitation Medicine.2019; 62(6): 435. CrossRef - Review: Clinical, genetic and neuroimaging features of frontotemporal dementia
R. Convery, S. Mead, J. D. Rohrer
Neuropathology and Applied Neurobiology.2019; 45(1): 6. CrossRef - Granulin in Frontotemporal Lobar Degeneration: Molecular Mechanisms of the Disease
Zemfira N. Karamysheva, Elena B. Tikhonova, Andrey L. Karamyshev
Frontiers in Neuroscience.2019;[Epub] CrossRef - The Whole Exome Sequencing Clarifies the Genotype- Phenotype Correlations in Patients with Early-Onset Dementia
Yangqi Xu, Xiaoli Liu, Junyi Shen, Wotu Tian, Rong Fang, Binyin Li, Jianfang Ma, Li Cao, Shengdi Chen, Guanjun Li, Huidong Tang
Aging and disease.2018; 9(4): 696. CrossRef - Mouse models of frontotemporal dementia: A comparison of phenotypes with clinical symptomatology
Rebekah M. Ahmed, Muireann Irish, Janet van Eersel, Arne Ittner, Yazi D. Ke, Alexander Volkerling, Julia van der Hoven, Kimi Tanaka, Tim Karl, Michael Kassiou, Jillian J. Kril, Olivier Piguet, Jürgen Götz, Matthew C. Kiernan, Glenda M. Halliday, John R. H
Neuroscience & Biobehavioral Reviews.2017; 74: 126. CrossRef - Translocator Protein-18 kDa (TSPO) Positron Emission Tomography (PET) Imaging and Its Clinical Impact in Neurodegenerative Diseases
Anne-Claire Dupont, Bérenger Largeau, Maria Santiago Ribeiro, Denis Guilloteau, Claire Tronel, Nicolas Arlicot
International Journal of Molecular Sciences.2017; 18(4): 785. CrossRef - Intrinsic functional connectivity alterations in progressive supranuclear palsy: Differential effects in frontal cortex, motor, and midbrain networks
Johannes Rosskopf, Martin Gorges, Hans‐Peter Müller, Dorothée Lulé, Ingo Uttner, Albert C. Ludolph, Elmar Pinkhardt, Freimut D. Juengling, Jan Kassubek
Movement Disorders.2017; 32(7): 1006. CrossRef - Phenotypic variability related to C9orf72 mutation in a large Sardinian kindred
Gianluca Floris, Giuseppe Borghero, Francesca Di Stefano, Rosanna Melis, Roberta Puddu, Laura Fadda, Maria R. Murru, Daniela Corongiu, Stefania Cuccu, Stefania Tranquilli, Antonino Cannas, Maria G. Marrosu, Adriano Chiò, Francesco Marrosu
Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration.2016; 17(3-4): 245. CrossRef - Clinical features ofTBK1carriers compared withC9orf72,GRNand non-mutation carriers in a Belgian cohort
Sara Van Mossevelde, Julie van der Zee, Ilse Gijselinck, Sebastiaan Engelborghs, Anne Sieben, Tim Van Langenhove, Jan De Bleecker, Jonathan Baets, Mathieu Vandenbulcke, Koen Van Laere, Sarah Ceyssens, Marleen Van den Broeck, Karin Peeters, Maria Mattheijs
Brain.2016; 139(2): 452. CrossRef - Characterization of Movement Disorder Phenomenology in Genetically Proven, Familial Frontotemporal Lobar Degeneration: A Systematic Review and Meta-Analysis
Carmen Gasca-Salas, Mario Masellis, Edwin Khoo, Binit B. Shah, David Fisman, Anthony E. Lang, Galit Kleiner-Fisman, Patrick Lewis
PLOS ONE.2016; 11(4): e0153852. CrossRef - The clinical spectrum of sporadic and familial forms of frontotemporal dementia
Ione O. C. Woollacott, Jonathan D. Rohrer
Journal of Neurochemistry.2016; 138(S1): 6. CrossRef - Genetics of Progressive Supranuclear Palsy
Sun Young Im, Young Eun Kim, Yun Joong Kim
Journal of Movement Disorders.2015; 8(3): 122. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite