 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 14(1); 2021 > Article
-
Case Report
New Nonsense Variant c.2983G>T; p.Glu995* in the CACNA1A Gene Causes Progressive Autosomal Dominant Ataxia -
Yannic Saathoff1
 , Saskia Biskup2,3, Claudia Funke3, Christian Roth1,4
, Saskia Biskup2,3, Claudia Funke3, Christian Roth1,4 -
Journal of Movement Disorders 2021;14(1):70-74.
DOI: https://doi.org/10.14802/jmd.20082
Published online: October 31, 2020
1Department of Neurology and Neurophysiology, DRK-Kliniken Nordhessen, Kassel, Germany
2Center for Genomics and Transcriptomics (CeGaT) GmbH, Tuebingen, Germany
3Practice for Human Genetics, Tuebingen, Germany
4Department of Neurology, University of Marburg, Marburg, Germany
- Corresponding author: Yannic Saathoff, MD, PhD Department of Neurology and Neurophysiology, DRK-Kliniken Nordhessen, Hansteinstraße 29, 34121 Kassel, Germany / Tel: +49 561 3086 2400 / Fax: +49 561 3086 2404
Copyright © 2021 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The genetic testing of hereditary ataxias includes screening for CAG-repeat expansions as well as pathogenic variants and nontranslated oligonucleotide expansion, which can cause spinocerebellar ataxia (SCA). Genotype-phenotype correlations of several SCA subtypes are difficult to establish, and the underlying mechanisms remain unclear. Here, we report a 58-year-old male patient who presented with severe generalized ataxia, horizontal gaze-evoked nystagmus, cognitive impairment and a positive family history of gait difficulties. Genetic panel diagnostics revealed a new nonsense pathogenic variant in the CACNA1A gene (c.2983G>T; p. Glu995*) that segregated with the phenotype in three clinically affected family members. This gene is related to SCA type 6 (SCA6), episodic ataxia type 2, familial hemiplegic migraine type 1, among others. When it is supported by the clinical findings and family history, additional DNA sequencing beyond fragment length analysis should be performed.
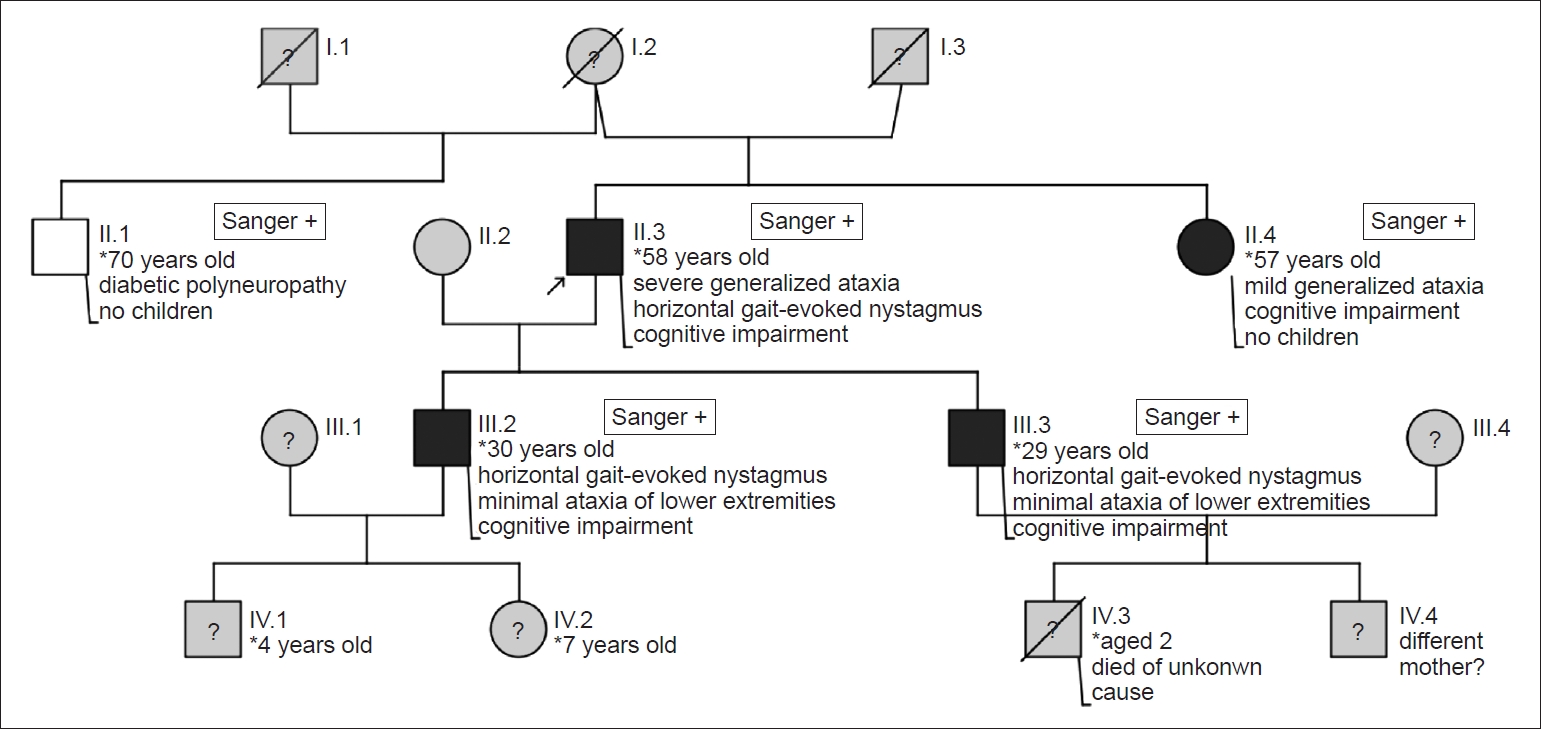
- Between 2018 and 2019, five family members were examined in our clinic (Department of Neurology, DRK-Kliniken Nordhessen, Kassel, Germany). After written informed consent was obtained, genetic analyses were performed in four clinically affected patients as well as in one unaffected family member. DNA was extracted from peripheral blood according to standard protocols. We initiated molecular genetic panel analyses of the index patient by investigating CAG repeats of the responsible genes for SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17, with negative results. Afterwards, initial sequence analysis was performed by using a panel-based next-generation sequencing (NGS) approach. The following genes were analyzed: ATM (NM_000051.3), APTX (NM_175073.2), SETX (NM_015046.6), ANO10 (NM_ 018075.4), PRKCG (NM_002739.4), and CACNA1A (NM_001127 221.1). After a new pathogenic nonsense variant c.2983G>T; p.Glu995* was identified in CACNA1A in the index patient, all other family members were tested for the identified variant according to a Sanger sequencing protocol. Informed consent for collecting and publishing the data was obtained from all of the examined patients. The study was approved by the Institutional Review Board at the University Medical Center Göttingen (approval no: 20/11/19).
- Patient 1 (index patient; patient II.3 in Figure 1)
- In 2018, a 58-year-old male patient presented with persistent pain in both knees in the Department of Pain Medicine of our clinic. He had lumbar spinal disk herniations that were treated by hemilaminectomy earlier that year. He required two additional surgeries afterwards due to discitis. He also complained of the progressive inability to lift both of his arms and has only been able to walk with a walker since 2015. The patient had a history of chronic cervicobrachialgia with weakness in both arms and underwent fusion as well as cervical spinal canal decompression the previous year. The surgery relieved the pain without affecting motor function. His medical history included mild hypertension and mild alcohol and non-steroidal anti-inflammatory drug abuse. The neurological exam revealed proximal weakness in both upper extremities, which he could not lift by more than 90 degrees; shoulder girdle atrophy including the pectoral and trapezoid muscles, saccades and horizontal gaze-evoked nystagmus on both sides; unsteady gait without assistance; and straddle-legged ataxia. The Babinski sign was positive bilaterally. In the detailed history taking, the patient reported progressive difficulties with walking for at least 10 years. He had been working as a furniture mover until 2001. His two children were reported to be healthy; however, his 54-year-old sister also needed a walker. Both parents died of an unknown cause long ago. A history of migraine was not reported.
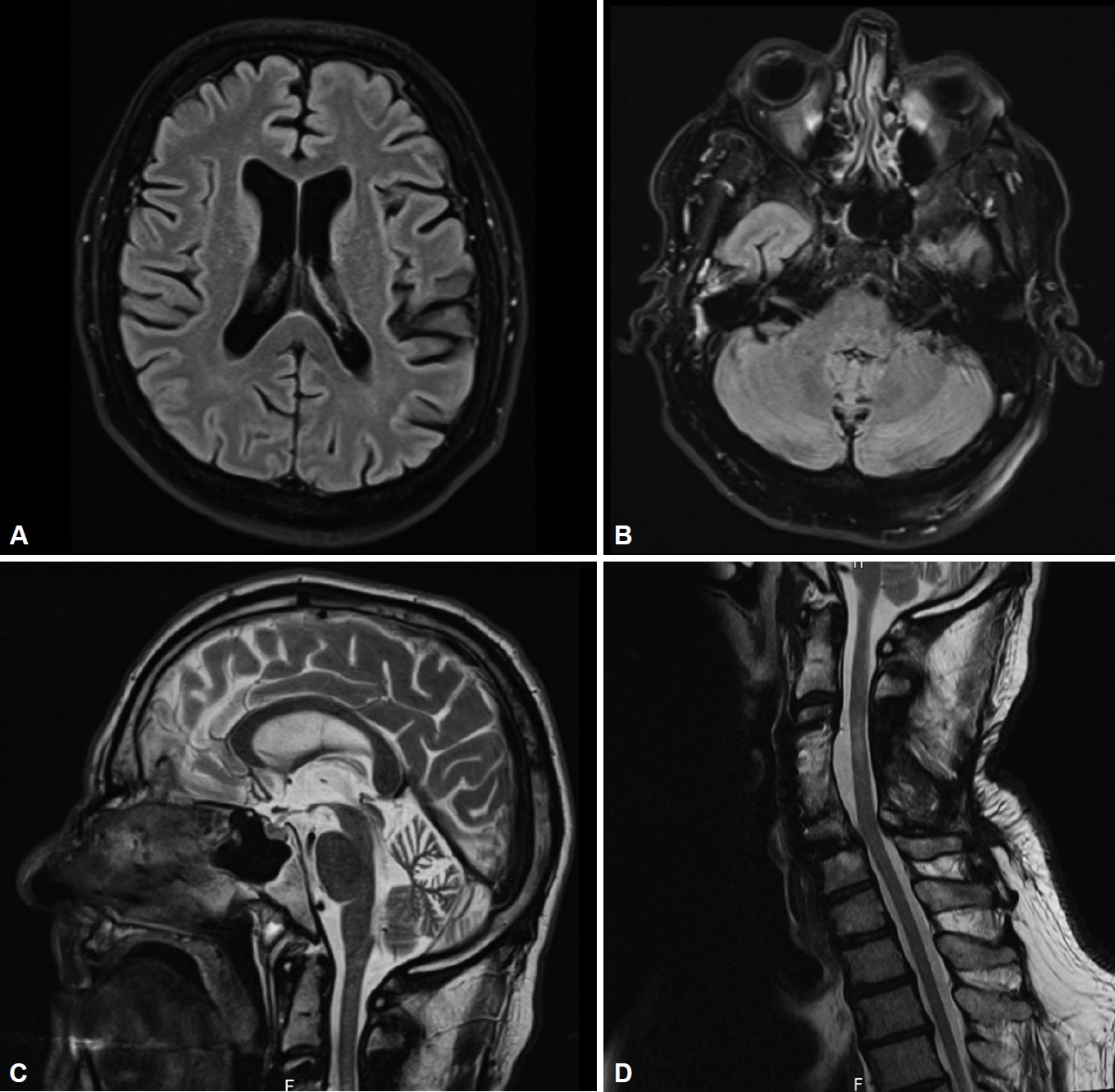
- The diagnostic tests included extensive laboratory blood and cerebrospinal fluid tests as well as MRI scans of the head and cervical spine, evoked potential examinations (sensory and motor) and electroneurography. Ultimately, the tests showed moderate creatine kinase elevation, slight generalized brain atrophy, vascular leukoencephalopathy, cervical myelopathy C3/C4 and the beginning stage of spinal canal stenosis at the level of C5/C6 on the MRI examinations (Figure 2). After excluding the common causes of ataxia, we initiated genetic panel testing. However, the results did not show polyglutamine expansions in the common SCA-associated genes for SCA1, SCA2, SCA3, SCA6, SCA7, or SCA17. Then, NGS revealed a nonsense variant c.2983G>T; p.Glu995* in the CACNA1A gene.
- Patients 2, 3, and 4 (patients II.4, III.2, and III.3 in Figure 1)
- The 57-year-old sister of the propositus presented with mild generalized ataxia, cognitive impairment, gait difficulties with an unsteady broad-legged gait and undirected falling tilt. Additionally, she showed pes cavus and claw toes on both sides. She required a walker for a couple of years due to the progressive worsening of her gait and had chronic back pain and osteoporosis. She had undergone multiple spine surgeries. Unfortunately, MRI scans could not be taken.
- The 30-year-old son of the propositus did not report any difficulties with walking or coordination or additional diseases, and he had two healthy children. The neurological exam revealed inexhaustible horizontal gaze-evoked nystagmus on both sides, a slight ataxic heel-knee test result and cognitive impairment. The MRI brain scan was unremarkable.
- The 29-year-old son of the propositus reported always being clumsy during sports in elementary school and had a legal guardian due to cognitive impairment. The clinical examination showed an exhaustible horizontal bilateral gaze-evoked nystagmus, a slight ataxic heel-knee test and cognitive impairment. No MRI scans were performed.
- Patients 2, 3, and 4 did not have a history of migraine or recurrent hemiparesis. Segregation analyses by Sanger sequencing confirmed the presence of the new nonsense variant c.2983G>T; p.Glu995* in CACNA1A in patients 2, 3, and 4.
- The half-brother of the propositus was 70 years old and had the same mother as the propositus. He had a history of hypertension and a known polyneuropathy due to insulin-dependent diabetes mellitus. The clinical exam showed mild clinical signs of polyneuropathy but no signs of ataxia or oculomotor disturbances. Sanger sequencing did not show the pathogenic variant in CACNA1A.
CASE REPORT
Patient 2
Patient 3
Patient 4
Patient 5 (patient II.1 in Figure 1)
- We identified a new nonsense variant c.2983G>T; p.Glu995* in the CACNA1A gene to be a cause of progressive ataxia and cognitive impairment. To the best of our knowledge, p.Glu995* in CACNA1A has not been described in the literature to date. The family showed an autosomal dominant pattern of inheritance. The nonsense variant most likely leads to a truncated protein or nonsense-mediated mRNA decay. Furthermore, all four family members with a confirmed variant showed signs of cerebellar syndrome. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, there is strong evidence for the pathogenicity of this new nonsense variant [1]. The pathogenicity of the variant was confirmed by an online genetic variant interpretation tool based on the ACMG guidelines (using PVS1, PS4, and PP1) [2].
- Interestingly, variants in the CACNA1A gene are associated with familial hemiplegic migraine type 1 (FHM1), episodic ataxia type 2 (EA2), and SCA6. The CACNA1A gene codes for the α1A subunit of the CaV2.1 or P/Q-type voltage-gated calcium channel [3]. A polyglutamine expansion in CACNA1A is the cause of SCA6, while in FMH1 pathogenic missense variants and in EA2 missense variants, deletions, insertions and splice site variants in CACNA1A have been found to be causative factors [4,5]. The mechanism that causes SCA6 is not fully understood. Different mechanisms have been proposed, but some of the reported results are conflicting [3,6]. There might also be a clinical overlap between SCA6, EA2, and FHM1 [4]. However, in the family in our study, none of the patients showed clinical signs of EA2 or FHM1.
- SCA6 is caused by CAG-repeat expansion in exon 47 of the CACNA1A gene. In SCA6, the number of CAG repeats does not have an effect on disease progression but instead affects the age of onset [7]. In 1997, Yue et al. [8] described a type of dominantly inherited ataxia caused by a variant in the CACNA1A gene. Recently, more variants in addition to the polyglutamine expansion in the CACNA1A gene have been detected: Coutelier et al. [9] defined channelopathies as point mutations in channel genes such as the CACNA1A, KCND3, and KCNC3, genes rather than CAG-repeat expansions in ataxia-linked genes. Here, we describe another point mutation, the pathogenic nonsense variant c.2983G>T; p.Glu995* in the CACNA1A gene that causes progressive ataxia.
- A previous study established a correlation between the type of variant and the age of onset of the disease as well as some of the clinical features [9]. The authors found that in patients with point mutations, the onset of the disease occurs at an earlier age, the disease progression is slower and the phenotype is slightly different (pure cerebellar syndrome, often combined with intellectual deficiency).
- However, in the family included in our study, patients 1 and 2 did not show an early onset of the disease, while the severity of cerebellar syndrome in patients 3 and 4 was mild. While the latter two patients did not complain about any disabilities, signs of ataxia were detected in our neurological examinations. Patient 1 and 3 also did not show prominent cerebellar atrophy, which Coutelier et al. [9] found to be especially present in patients with CACNA1A point-mutated ataxias. Cognitive impairment was indeed present in all four affected patients. Even when there is a difference between the genotype-phenotype correlations, it is still important to distinguish between polyglutamine expansion ataxias and non-polyQ repeat ataxias since patients with the former ataxias have a reported shorter survival [10].
- In summary, we found a previously undescribed nonsense variant in the CACNA1A gene that causes progressive autosomal dominant ataxia. When classic fragment length analyses do not show CAG repeat expansions, additional genetic testing such as NGS should be performed to screen for additional pathogenic variants, especially in cases of hereditary ataxias. The clinical differentiation between channelopathies and polyQ ataxias is difficult. The patient history needs to be taken carefully to screen for other possible phenotypes of a variant in CACNA1A, such as episodic ataxia or familial hemiplegic migraine.
DISCUSSION
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Christian Roth. Data curation: Yannic Saathoff. Investigation: Yannic Saathoff, Christian Roth, Claudia Funke, Saskia Biskup. Methodology: Yannic Saathoff, Christian Roth. Project administration: Christian Roth, Yannic Saathoff. Resources: Christian Roth, Saskia Biskup. Software: Yannic Saathoff. Supervision: Christian Roth. Validation: Yannic Saathoff, Christian Roth, Claudia Funke. Visualization: Yannic Saathoff. Writing—original draft: Yannic Saathoff. Writing—review & editing: Yannic Saathoff, Christian Roth, Claudia Funke, Saskia Biskup.
Notes
- None.
Acknowledgments
- 1. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.ArticlePubMedPMC
- 2. Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med 2016;18:1165.ArticlePubMedPMC
- 3. Solodkin A, Gomez CM. Spinocerebellar ataxia type 6. Handb Clin Neurol 2012;103:461–473.ArticlePubMed
- 4. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007;130:2484–2493.ArticlePubMed
- 5. Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology 2004;62:17–22.ArticlePubMed
- 6. Giunti P, Mantuano E, Frontali M, Veneziano L. Molecular mechanism of Spinocerebellar Ataxia type 6: glutamine repeat disorder, channelopathy and transcriptional dysregulation. The multifaceted aspects of a single mutation. Front Cell Neurosci 2015;9:36.ArticlePubMedPMC
- 7. Jacobi H, du Montcel ST, Bauer P, Giunti P, Cook A, Labrum R, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14:1101–1108.ArticlePubMed
- 8. Yue Q, Jen JC, Nelson SF, Baloh RW. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am J Hum Genet 1997;61:1078–1087.ArticlePubMedPMC
- 9. Coutelier M, Coarelli G, Monin ML, Konop J, Davoine CS, Tesson C, et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 2017;140:1579–1594.ArticlePubMed
- 10. Monin ML, Tezenas du Montcel S, Marelli C, Cazeneuve C, Charles P, Tallaksen C, et al. Survival and severity in dominant cerebellar ataxias. Ann Clin Transl Neurol 2015;2:202–207.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- The genotype–phenotype correlations of the CACNA1A-related neurodevelopmental disorders: a small case series and literature reviews
Miriam Kessi, Baiyu Chen, Nan Pang, Lifen Yang, Jing Peng, Fang He, Fei Yin
Frontiers in Molecular Neuroscience.2023;[Epub] CrossRef - Next-Generation Sequencing Technologies and Neurogenetic Diseases
Hui Sun, Xiao-Rong Shen, Zi-Bing Fang, Zong-Zhi Jiang, Xiao-Jing Wei, Zi-Yi Wang, Xue-Fan Yu
Life.2021; 11(4): 361. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

