 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 14(1); 2021 > Article
-
Case Report
Sialidosis Type I without a Cherry Red Spot— Is There a Genetic Basis? -
Koti Neeraja1*
 , Vikram Venkappayya Holla1*, Shweta Prasad1,2, Bharath Kumar Surisetti1, Kempaiah Rakesh1, Nitish Kamble1, Ravi Yadav1, Pramod Kumar Pal1
, Vikram Venkappayya Holla1*, Shweta Prasad1,2, Bharath Kumar Surisetti1, Kempaiah Rakesh1, Nitish Kamble1, Ravi Yadav1, Pramod Kumar Pal1 -
Journal of Movement Disorders 2021;14(1):65-69.
DOI: https://doi.org/10.14802/jmd.20083
Published online: October 31, 2020
1Department of Neurology, National Institute of Mental Health and Neurosciences, Karnataka, India
2Department of Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Karnataka, India
- Corresponding author: Pramod Kumar Pal, MD, DNB, DM, FRCP Department of Neurology, National Institute of Mental Health and Neurosciences (NIMHANS), Hosur Road, Bengaluru-560029, Karnataka, India / Tel: +91-80-26995147 / Fax: +91-80-26564830 / E-mail: palpramod@hotmail.com
- *This authors contributed equally to this work.
Copyright © 2021 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Sialidosis is an inborn error of metabolism due to a defect in the NEU1 gene and manifests as two phenotypes: mild type I and severe type II. The cherry red spot (CRS) is a characteristic feature in both types of sialidosis; reports of sialidosis without a CRS are rare. We report two cases of genetically confirmed sialidosis type I with a typical presentation of progressive cortical myoclonus and ataxia but without the CRS. A previously reported homozygous pathogenic variant p.Arg294Cys was detected in the first case, and a novel homozygous pathogenic variant p.Arg305Pro was detected in the second case. Additionally, we reviewed the literature describing cases with similar mutations to find a genetic basis for the absence of a CRS. Milder mutation of both alleles detected in both patients may be the reason for the absence of a CRS.
- Case 1
- A 33-year-old man presented with 14-year history of progressive shaking of the left upper limb (UL) followed by the right UL that 3 years later involved both lower limbs (LL). Shaking was predominantly action-induced, jerky, and led to occasional dropping of objects. The patient had difficulty walking and gait initiation along with an imbalance and falls. He also reported slurring of speech with no other bulbar symptoms. He had no significant family history. At presentation, the patient was independent, employed, and had comprehensible speech. A video was recorded after a written informed consent was obtained.
- On examination, cognition was normal. Ophthalmological examination by a neuro-ophthalmologist was normal, including acuity, anterior segment and fundus examination. He had bilateral horizontal gaze-evoked nystagmus, mild cerebellar dysarthria, appendicular and axial incoordination, and ataxic gait. He had distal predominant, action-induced, multifocal myoclonus involving all four limbs with no sensory or auditory sensitivity. Upon standing, there were tremulous movements of both LL with an ataxic gait (Supplementary Video 1 in the online-only Data Supplement). The rest of the examination was normal. He was considered to be a case of progressive myoclonus ataxia (PMA) and was evaluated accordingly.
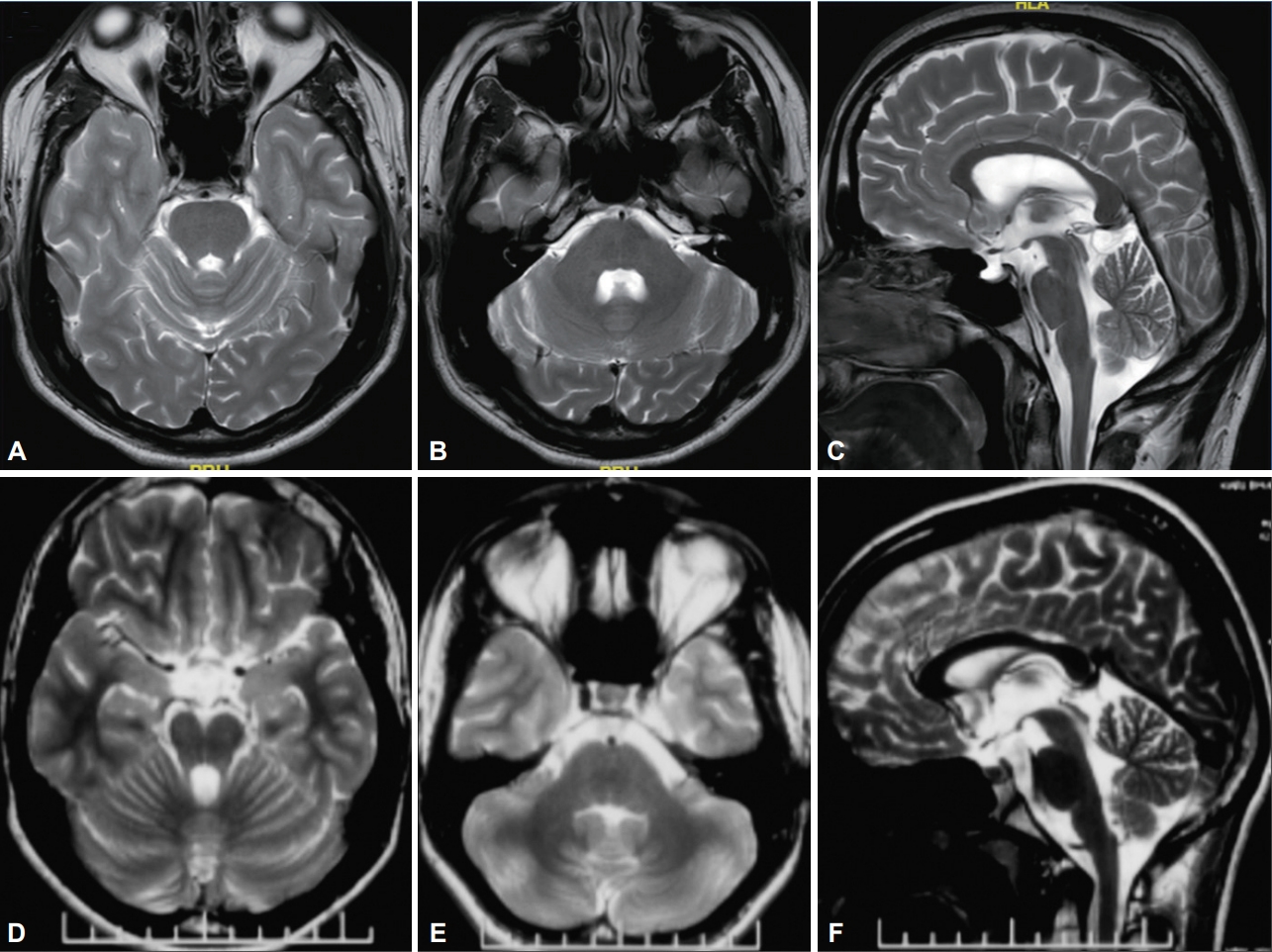
- The routine blood investigations, ultrasound of the abdomen, and MRI of the brain were normal. Electrophysiological evaluation revealed giant somatosensory evoked potentials (SSEP) with N20–P25 amplitude of 14 µV (normal value < 10 µV), enhanced long loop reflexes, and frequent quasi-rhythmic bursts of < 50 ms duration in all four limbs according to the surface electromyography (EMG) suggesting the cortical nature of the myoclonus. Additionally, visually evoked potentials (VEP) showed bilaterally prolonged P100 latencies of 112 ms on the right eye and 114 ms on the left eye (normal value 106 ± 4 ms). However, electroencephalogram (EEG) and nerve conductivity study (NCS) were normal. Clinical exome sequencing revealed a previously reported pathogenic homozygous missense variant in exon 5 of the NEU1 gene (chr6: g.31827960G> A; p.Arg294Cys; ENST 00000375631.4) [4,5]. Based on the clinical and genetic data, the patient was diagnosed with sialidosis type I and was treated with clonazepam, sodium valproate, and levetiracetam. He had mild improvement in myoclonus, gait, and falls.
- Case 2
- A 14-year-old boy with a second-degree consanguineous parentage presented with one-year history of tremulousness in both ULs, which was postural, jerky, and affected his daily activities. He also had a mild imbalance while walking with difficulty negotiating narrow passages and had difficulty riding a bicycle. He had a history of decreased hearing in the left ear. There was no significant family history pertaining to his illness. The video was taken after a written informed consent was obtained.
- On examination, the patient had mild deficits in executive function, verbal memory, and verbal fluency. Ophthalmological examination by a neuro-ophthalmologist was normal, including acuity, anterior segment, and fundus examination. There were hypermetric saccades, jerky pursuits, bilateral gaze-evoked nystagmus, and sensorineural hearing impairment in the left ear. There were subtle, distal predominant, postural and actioninduced myoclonic jerks in ULs (Supplementary Video 2 in the online-only Data Supplement). Appendicular incoordination and wide-based and ataxic gait with impaired tandem was also noted (Supplementary Video 2 in the online-only Data Supplement). The rest of the neurological and other system examinations were normal. Based on these findings, PMA was suspected, and he was evaluated.
- The routine investigations including plasma ammonia and plasma lactate, and ultrasound of the abdomen was performed and were normal. Pure tone audiometry revealed mild bilateral sensorineural hearing loss. Electrophysiological evaluation revealed giant SSEP with N20–P25 amplitude of 34 µV (normal value < 10 µV), enhanced long loop reflexes, and frequent quasirhythmic bursts of < 50 ms duration in all four limbs according to the surface EMG suggesting cortical nature of the myoclonus. Bilateral VEP had prolonged P100 latencies of 128 ms on the right eye and 131 ms on the left eye (normal value 106 ± 4 ms). EEG and NCS were normal. The clinical exome sequencing revealed a novel homozygous missense variant in exon 5 of the NEU1 gene (chr6: g.31827926C>G; p.Arg305Pro; ENST 00000375631.4). This variant was classified as likely pathogenic according to the American College of Medical Genetics guidelines (PM2, PM5, PP2, and PP3) [2]. The final diagnosis of sialidosis type I was made based on these observations.
CASE REPORT
- The CRS is a characteristic feature of sialidosis, which is a lysosomal storage disorder caused by alpha-N-acetyl NEU deficiency secondary to NEU1 gene mutations. In the present report, both cases had an onset of the symptoms in the second decade with cortical myoclonus followed by ataxia and without dysmorphism or organomegaly, i.e., a sialidosis type I presentation. However, contrary to the usual description of a cherry red spot-myoclonus syndrome, both patients lacked a CRS [1]. Interestingly, the patients did not have a CRS or visual disturbances; however, VEP was prolonged in both patients. To the best of our knowledge, this is the first report of sialidosis type I without a CRS from India.
- More than 40 different mutations have been reported in the NEU1 gene; most mutations are missense. Diverse clinical phenotypes result from the genotypic heterogeneity. Manifestations of the mutations of the NEU1 gene vary from mild to severe. Genotype-phenotype correlation studies suggested that residual NEU activity is present in mild mutations; however, the activity is absent in severe mutations leading to type II phenotype [6]. To explain the phenotypic characteristics of two cases described in the present report (especially the absence of a CRS) based on their genotype, we analyzed the previously reported cases having either an identical amino acid substitution or a different amino acid substitution in the same locus (Table 1).
- Our first case had a homozygous mutation of the NEUI gene resulting in the p.Arg294Cys amino acid substitution. This mutation has been previously described in two case reports, including a homozygous case and a compound heterozygous case. The case with homozygous p.Arg294Cys mutation had a phenotype similar to that observed in our study, i.e., progressive myoclonus and ataxia without a CRS and seizures [4]. However, unlike our case, there was cognitive impairment, reduced visual acuity, nyctalopia, latent squint, skew deviation, mild thickening in perifoveal macula on optical coherence tomography (OCT), and cerebellar atrophy according to MRI of the brain. Another case with compound heterozygous mutation of the NEUI gene had a p.Arg294Cys mutation in one allele and a more severe truncating frame shift mutation in the other allele resulting in a typical type I phenotype, i.e., progressive ataxia, myoclonus, and a CRS [5]. Two cases of sialidosis type I with a CRS were reported by Bonten et al. [6] to have a different amino acid substitution at the same locus (p.Arg294Ser). However, in both cases, there was an additional severe mutation of the other allele in compound heterozygous state. A review of previously reported genetically validated cases of sialidosis type I described 20 of 21 cases without a CRS as having a combination of 4 missense variants (p.Ser182Gly, p.Gly227Arg, p.R305C, and p.S67I) [7]. Fourteen patients had homozygous p.Ser182Gly variant, 3 patients had homozygous p.Ser67Ile variant, 2 patients had compoundheterozygous p.Gly227Arg/p.Arg305Cys variants, and 1 patient had compound-heterozygous p.Ser182Gly/p.Gly227Arg variants [2,3,7]. Hence, it is possible that milder mutations of both alleles may lead to a type I phenotype without a CRS and a severe mutation of one of the alleles may lead to a type I phenotype with a CRS. The slow rate of progression and lower severity of the illness in our first case, who had a 14-year-long duration of the illness, supports the relatively benign nature of this mutation (p.Arg294Cys).
- Our second case had a novel homozygous p.Arg305Pro amino acid substitution. A different amino acid substitution in the same locus (p.Arg305Cys) has been reported to result in a type I phenotype similar to the phenotype observed in our case, which was characterized by myoclonus, ataxia, no seizures, normal acuity, no CRS, and enhanced SSEP but with a later age at onset, normal VEP, and normal MRI of the brain. However, it was a compound heterozygous configuration with a different missense mutation (p.Gly227Arg) (Table 1) [2]. A CRS may sometimes appear later in the course of the illness when the deposition of sialylated glycoproteins is increased [8]; since our second case was examined within a year of the onset of the illness, a prolonged follow up is required. OCT and autoflourescence examination of the fundus are more sensitive and can demonstrate retinal changes even when the fundus examination is otherwise normal [9]. Additionally, a CRS may become less conspicuous with disease progression because ganglion cells die leading to the loss of distinction between the fovea and the surrounding region [10]. Environmental factors, diet, and genetic factors in addition to NEU mutations may play a role in the phenotypic heterogeneity [6].
- In conclusion, although the CRS has been described as a characteristic finding in sialidosis, it may be absent in cases of sialidosis type I with milder mutations of both alleles. Therefore, the absence of a CRS should not preclude a diagnosis of sialidosis in patients who present with PMA syndrome.
DISCUSSION
Supplementary Material
Video 1.
Video 2.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Koti Neeraja, Vikram Venkappayya Holla, Pramod Kumar Pal. Data curation: Vikram Venkappayya Holla, Koti Neeraja, Shweta Prasad, Bharath Kumar Surisetti, Kempaiah Rakesh. Supervision: Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Writing—original draft: Koti Neeraja, Vikram Venkappayya Holla, Kempaiah Rakesh. Writing—review & editing: Shweta Prasad, Bharath Kumar Surisetti, Nitish Kamble, Ravi Yadav, Pramod Kumar Pal.
Notes
- None.
Acknowledgments
| Study | Cases | Variant | Cherry red spot | VEP (P100 latency) | Vision |
|---|---|---|---|---|---|
| Present study | Case 1 | c.880C>T; p.Arg294Cys | Absent | Prolonged | Normal |
| c.880C>T; p.Arg294Cys | |||||
| Case 2 | c.914G>C; p.Arg305Pro | Absent | Prolonged | Normal | |
| c.914G>C; p.Arg305Pro | |||||
| Bou Ghannam et al. [4] | One | c.880C>T; p.Arg294Cys* | Absent | NA | Affected |
| c.880C>T; p.Arg294Cys* | |||||
| Ranganath et al. [5] | One | c.880C>T; p.Arg294Cys* | Present | NA | Normal |
| c.1191delG; p.Arg397fs | |||||
| Bonten et al. [6] | Two | c.878C>T; p.Arg294Ser* | Present | NA | Normal |
| c.690T>A; p.Leu231His | |||||
| c.898C>T; p.Arg294Ser* | Present | NA | Affected | ||
| c.654G>A; p.Gly218Ala | |||||
| Canafoglia et al. [2] | Three | c.913C>T; pArg305Cys* | Absent | Normal | Affected |
| c.679G>A; pGly227Arg | |||||
| c.913C>T; pArg305Cys* | Absent | Normal | Normal | ||
| c.679G>A; pGly227Arg | |||||
| c.913C>T; pArg305Cys* | Absent | Normal | Normal | ||
| c.679G>A; pGly227Arg |
- 1. Khan A, Sergi C. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics (Basel) 2018;8:29.ArticlePubMedPMC
- 2. Canafoglia L, Robbiano A, Pareyson D, Panzica F, Nanetti L, Giovagnoli AR, et al. Expanding sialidosis spectrum by genome-wide screening: NEU1 mutations in adult-onset myoclonus. Neurology 2014;82:2003–2006.ArticlePubMed
- 3. Lai SC, Chen RS, Wu Chou YH, Chang HC, Kao LY, Huang YZ, et al. A longitudinal study of Taiwanese sialidosis type 1: an insight into the concept of cherry-red spot myoclonus syndrome. Eur J Neurol 2009;16:912–919.ArticlePubMed
- 4. Bou Ghannam AS, Mehner LC, Pelak VS. Sialidosis type 1 without cherryred spot. J Neuroophthalmol 2019;39:388–390.ArticlePubMed
- 5. Ranganath P, Sharma V, Danda S, Nandineni MR, Dalal AB. Novel mutations in the neuraminidase-1 (NEU1) gene in two patients of sialidosis in India. Indian J Med Res 2012;136:1048–1050.PubMedPMC
- 6. Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet 2000;9:2715–2725.ArticlePubMed
- 7. Ahn JH, Kim AR, Lee C, Kim NKD, Kim NS, Park WY, et al. Type 1 sialidosis patient with a novel deletion mutation in the NEU1 gene: case report and literature review. Cerebellum 2019;18:659–664.ArticlePubMed
- 8. Palmeri S, Villanova M, Malandrini A, van Diggelen OP, Huijmans JG, Ceuterick C, et al. Type I sialidosis: a clinical, biochemical and neuroradiological study. Eur Neurol 2000;43:88–94.ArticlePubMed
- 9. Han X, Wu S, Wang M, Li H, Huang Y, Sui R. Genetic and clinical characterization of mainland Chinese patients with sialidosis type 1. Mol Genet Genomic Med 2020;8:e1316.ArticlePubMedPMC
- 10. Nakaya-Onishi M, Suzuki A, Okamoto N, Fukada M. Observations on time course changes of the cherry red spot in a patient with Tay-Sachs disease. Br J Ophthalmol 2000;84:1318–1318.Article
REFERENCES
Figure & Data
References
Citations
- A fuzzy rule based machine intelligence model for cherry red spot disease detection of human eyes in IoMT
Kalyan Kumar Jena, Sourav Kumar Bhoi, Debasis Mohapatra, Chittaranjan Mallick, Kshira Sagar Sahoo, Anand Nayyar
Wireless Networks.2023; 29(1): 247. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
