E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 8(3); 2015 > Article
-
Case Report
Creutzfeldt-Jakob Disease in a Tertiary Care Hospital in Thailand: A Case Series and Review of the Literature - Praween Lolekha1,2, Ahmed Rasheed1, Chutanat Yotsarawat1
-
Journal of Movement Disorders 2015;8(3):136-140.
DOI: https://doi.org/10.14802/jmd.15014
Published online: September 10, 2015
1Neurology Division, Department of Internal Medicine, Faculty of Medicine, Thammasat University, Pathumthani, Thailand
2Stroke and Neurodegenerative Diseases Research Unit, Faculty of Medicine, Thammasat University, Pathumthani, Thailand
- Corresponding author: Praween Lolekha, MD, MSc, Neurology Division, Department of Internal Medicine, Faculty of Medicine, Thammasat University, 95 Moo 8 Paholyotin Road, Amphur Klongluang, Pathumthani 12120, Thailand / Tel: +66-2-9269794 / Fax: +66-2-9269793 / E-mail: pwlolekha@gmail.com
• Received: April 16, 2015 • Revised: May 11, 2015 • Accepted: June 30, 2015
Copyright © 2015 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Creutzfeldt-Jakob Disease (CJD) is an incurable and inevitably fatal neurodegenerative disorder. Although CJD has a worldwide distribution, there are no official statistics on CJD in Thailand. A diagnosis of CJD is suspected when a patient develops rapidly progressive dementia with myoclonus. However, CJD may be mistaken for a variety of illnesses because its initial presentation frequently consists of non-specific symptoms. Here, we examined cases of sporadic CJD (sCJD) from Thammasat University Hospital (a tertiary care hospital in Thailand) between January 1, 2012 and December 31, 2014. Three cases of probable and possible sCJD were collected. All cases presented with rapidly progressive cognitive dysfunction accompanied by spontaneous myoclonus. Classical electroencehalography changes and typical abnormal MRI features were observed. All of the cases died within a period of 8 months. None of the patients underwent brain biopsy. Our findings raise questions about the prevalence of CJD in Thailand, which needs further study.
- Case 1
- A 42-year-old Thai woman presented with sub-acute dizziness and an unsteady gait. Two weeks later, she developed memory impairment and deterioration of concentration. Her interest and pleasure in all activities, as well as her speech, diminished remarkably. Her condition became progressively worse, and eventually she became akinetic-rigid and mute. Later, spontaneous myoclonic jerks were present in her distal limbs. Magnetic resonance imaging (MRI) showed high signal intensity in the bilateral caudate nucleus, putamen and left frontotemporal cortex on T2-weighted and diffusion-weighted imaging (DWI). Electroencephalography (EEG) revealed generalized and synchronous periodic sharp wave complexes occurring at intervals of 0.6–0.8 seconds. A back-averaging EEG confirmed cortical origin multifocal myoclonus. She died 32 weeks after the initial presentation. Her clinical history together with physical signs, typical EEG, and brain MRI findings were consistent with a diagnosis of probable sCJD [1].
- Case 2
- A 76-year-old Thai man presented with a three-week history of confusion, hallucinations, dysarthria, and mild left-sided weakness. He denied a history of headache or fever. His general blood chemistry and cerebrospinal fluid (CSF) studies were within a normal range. A brain MRI study showed symmetric hypersignal intensity on fluid-attenuated inversion recovery (FLAIR) MRI sequences with restricted diffusion on DWI in his bilateral caudate nuclei, putamen and bilateral fronto-temporo-parietal cortices. An EEG revealed periodic generalized sharp wave complexes. His clinical status gradually deteriorated with time, and he developed spontaneous generalized myoclonic jerks. He developed aspiration pneumonia and passed away, succumbing to the illness 8 months after its onset. The patient was diagnosed as having probable sCJD [1].
- Case 3
- A 53-year-old Thai female teacher presented with a history of dizziness, fatigue, and insomnia. One month later, she developed social withdrawal, psychomotor retardation, and memory impairment. On physical examination, she was alert but had markedly decreased speech output. Generalized rigidity and occasional spontaneous myoclonic jerks were observed. A tentative diagnosis of akinetic-rigid syndrome was made, and full blood examinations and CSF investigations were performed. A computed tomography (CT) brain scan was unremarkable. An EEG revealed an intermittent delta slow wave superimposed on a normal alpha background. Her symptoms rapidly deteriorated, and she became bedridden and mute. Multiple spontaneous limb myoclonic jerks were observed. She died 4 months after symptom onset. Her clinical history, progression and physical findings suggested a diagnosis of possible sCJD [1].
CASE REPORT
- CJD is a human prion disease with characteristic clinical and diagnostic features. This fatal neurological disease occurs in sporadic, familial and acquired forms. sCJD is the most common type of CJD, accounting for at least 85 percent of cases [2]. In Thailand, there is limited literature on CJD, as it has been underreported and misdiagnosed. There is no regional or national surveillance system for CJD in Southeast Asia [3]. Furthermore, knowledge of this rare disease is limited to medical specialists. These facts reflect the need for public awareness, an effective reporting system, advanced medical education, and laboratory, neurological and neuropathological diagnostic capacity in this region. A literature search on the epidemiology of CJD in Southeast Asia revealed less than 25 published cases since the year 2000. There were no reports of variant, iatrogenic or familial CJD in this region [4,5]. CJD may be mistaken for a variety of illnesses because it typically initially presents as non-specific symptoms, such as dizziness, vertigo, insomnia, and personality and mood changes.
- CJD has a degree of clinicopathological heterogeneity. Recently, six different molecular subtypes of sCJD have been identified, which vary with respect to age of onset, disease duration, presenting symptoms, neuropathology, and MRI signal alterations [6]. In general, the average age of onset and length of illness in patients with sCJD are 60 years and 4 months, respectively [2]. However, younger patients trend toward having prolonged survival times [7]. Cerebellar and vestibular features are common and can be the only presenting symptoms, especially in young patients, resulting in misdiagnosis [7,8]. Cognitive decline and myoclonus may be delayed for weeks or even months after disease onset, causing difficulty in diagnosis.
- CJD can present as a movement disorder, and myoclonus is the most common abnormal movement that could occur at any stage; it can even be an initial manifestation of the illness. Myoclonic jerks are often generalized, asymmetric, cause greater effects to the distal extremities, and are associated with periodic sharp wave EEG activity [9]. Other movement disorders, including dystonia, choreoathetosis, tremor, hemiballismus, and atypical parkinsonian syndromes, have been described in a number of patients; however, these movements are usually combined with myoclonus [9]. Choreoathetosis is rare in sCJD but has been frequently reported in variant CJD (vCJD) and is listed in the diagnostic criteria for possible vCJD [10]. Atypical parkinsonian features, such as gait disorders, alien limb, and supranuclear gaze palsy, may occur as an initial presentation of CJD. Nevertheless, subacute progression rapidly combined with cognitive decline and myoclonus is suggestive of CJD [9]. Akinetic mutism (AM) is characterized by a marked reduction in motor function, including facial expression, gesture, and speech output, with preserved awareness. AM usually occurs in later stages of the disease and is considered a symptom that helps to establish the diagnosis of sCJD [1].
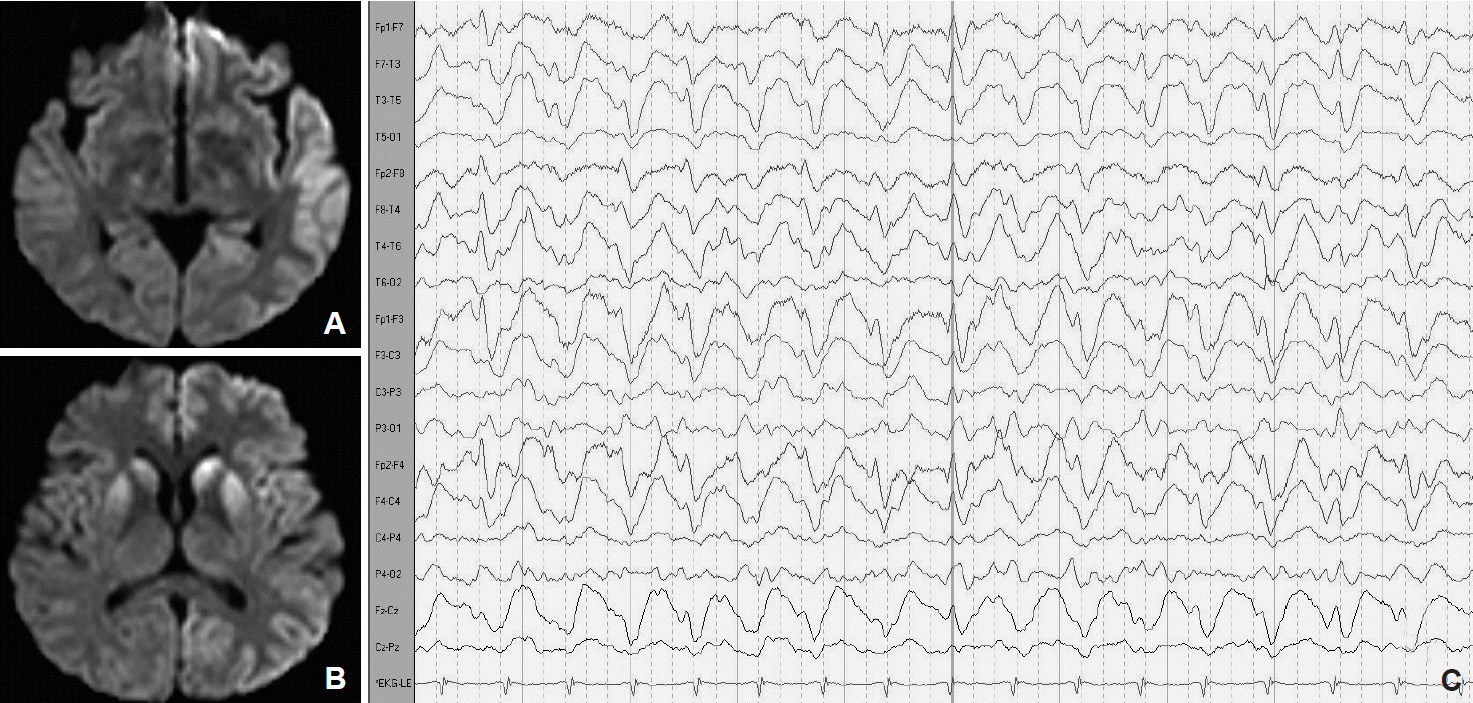
- A definite diagnosis of CJD requires brain tissue examination, which is not performed routinely by many institutions due to the transmissible nature of the disease. Therefore, diagnostic tools such as EEG and assays of certain brain proteins in CSF have been used to define probable cases of CJD [3]. Recent studies using MRI have shown its potential as a diagnostic tool in sCJD, showing a sensitivity and specificity of 98% and 94%, respectively [1]. These changes comprise alterations on DWI and FLAIR MRI sequences in the caudate nucleus and putamen or in at least two cortical regions (temporal, parietal, and occipital) (Fig. 1A and B) [1]. An abnormality on DWI-MRI could be the first diagnostic clue of CJD and could be detected as early as 3 weeks after symptom initiation and even before the appearance of an abnormal EEG [11]. MRI can also be used to differentiate sCJD from vCJD. In vCJD, abnormalities are commonly located in the posterior and medial thalami followed by the periaqueductal grey matter, striatum, and less commonly the neo-cortex. An increased signal intensity in the posterior nuclei of the thalamus is known as a pulvinar sign and is the most sensitive marker for vCJD [11].
- Several studies have demonstrated the benefit of evaluating 14-3-3 proteins in CSF for the diagnosis of sCJD [12]. However, an elevation of 14-3-3 proteins is not sensitive and specific for CJD. A number of other conditions that cause extensive neuronal damage, such as stroke, subarachnoid hemorrhage, hypoxic brain damage, encephalitis, and paraneoplastic encephalopathy, may also give positive 14-3-3 CSF results [3]. Another method is the use of a ratio of CSF total tau versus phosphorylated tau, which raised diagnostic specificity by up to 99%, but sensitivity remained relatively low at 79% [13]. Recently, the use of real-time quaking-induced conversion analysis of nasal brushings from olfactory epithelium has demonstrated a sensitivity of 97% and a specificity of 100% for the detection of CJD [14]. Unfortunately, these protein analysis assays are not available in Thailand.
- EEG is an important diagnostic tool for the diagnosis of CJD. Approximately 60–80% of cases are reported to develop a characteristic appearance of 0.5–2 Hz periodic, generalized bursts of spike wave complexes (Fig. 1C). Typical EEG findings may be absent during the initial stages but often become apparent as the disease progresses. In most cases, abnormal EEG findings occur approximately 12 weeks after clinical onset. In our case series, characteristic EEG findings were found in 2 cases at 6 weeks after clinical onset, and they persisted to appear as abnormal at the end stage of the disease.
- In conclusions, CJD is clinically heterogeneous and has a wide range of presenting symptoms. We report 3 cases with probable and possible sCJD presenting with non-specific clinical features and rapidly developed dementia, personality changes, parkinsonism culminating in AM and myoclonus during a 3-year period. EEG and MRI are important diagnostic tools that can detect abnormalities in early stages of the disease. Our findings raise questions about the true prevalence of CJD in Thailand, which needs further research and national surveillance. A definite and accurate diagnosis is crucial for patients, patients’ families and healthcare providers for preventing transmission and also for community surveillance of this infectious and transmissible condition.
DISCUSSION
- We thank the patients and their families for their cooperation in this report.
Acknowledgments
Figure 1.Diffusion-weighted magnetic resonance imaging showing increased signal intensity in the left frontotemporal cortex (A) and bilateral caudate nucleus and putamen (B). Periodic generalized sharp wave complexes detected by electroencehalography (C).
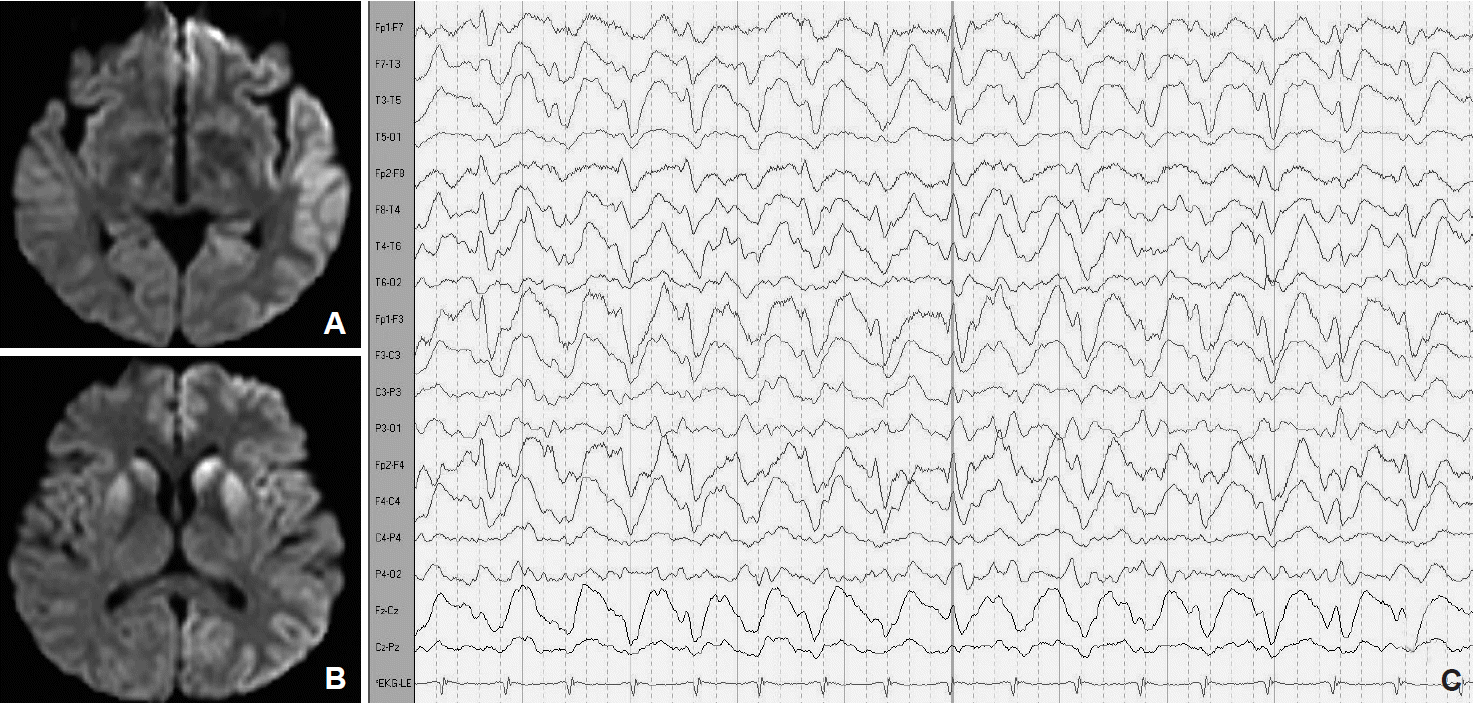
Table 1.Clinical presentations and investigations of cases
- 1. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009;132(Pt 10):2659–2668.ArticlePubMedPMCPDF
- 2. Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994;35:513–529.ArticlePubMed
- 3. Global surveillance, diagnosis and therapy of human Transmissible Spongiform Encephalopathies: report of a WHO consultation. Geneva, Switzerland: WHO; 1998.
- 4. Kandiah N, Tan K, Pan AB, Au WL, Venketasubramanian N, Tchoyoson Lim CC, et al. Creutzfeldt-Jakob disease: which diffusion-weighted imaging abnormality is associated with periodic EEG complexes? J Neurol 2008;255:1411–1414.ArticlePubMed
- 5. Law ZK, Subramaniam SR, Tan HJ, Azmin S, Osman SS, Nafisah WN, et al. Creutzfeldt-Jakob disease: a first case series from a tertiary hospital in malaysia and review of literature in Southeast Asia. Clin Res Infect Dis 2014;1:1008.
- 6. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull 2003;66:213–239.ArticlePubMedPDF
- 7. Appleby BS, Appleby KK, Rabins PV. Does the presentation of Creutzfeldt-Jakob disease vary by age or presumed etiology? A meta-analysis of the past 10 years. J Neuropsychiatry Clin Neurosci 2007;19:428–435.ArticlePubMed
- 8. Corato M, Cereda C, Cova E, Ferrarese C, Ceroni M. Youngonset CJD: age and disease phenotype in variant and sporadic forms. Funct Neurol 2006;21:211–215.PubMed
- 9. Maltête D, Guyant-Maréchal L, Mihout B, Hannequin D. Movement disorders and Creutzfeldt-Jakob disease: a review. Parkinsonism Relat Disord 2006;12:65–71.ArticlePubMed
- 10. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000;47:575–582.ArticlePubMed
- 11. Meissner B, Körtner K, Bartl M, Jastrow U, Mollenhauer B, Schröter A, et al. Sporadic Creutzfeldt-Jakob disease: magnetic resonance imaging and clinical findings. Neurology 2004;63:450–456.ArticlePubMed
- 12. Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:811–815.ArticlePubMed
- 13. Skillbäck T, Rosén C, Asztely F, Mattsson N, Blennow K, Zetterberg H. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol 2014;71:476–483.ArticlePubMed
- 14. Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, Groveman BR, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med 2014;371:519–529.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Sporadic Creutzfeldt-Jakob disease: Brain MRI lesion features from 2 cases reports
Hoang Dinh Au, Nguyen Thu Lan, Nguyen Thai Binh, Le Tuan Linh, Ma Mai Hien, Nguyen Minh Duc
Radiology Case Reports.2024; 19(3): 939. CrossRef - A systemic analysis of Creutzfeldt Jakob disease cases in Asia
Urwah Rasheed, Sana Khan, Minahil Khalid, Aneeqa Noor, Saima Zafar
Prion.2024; 18(1): 11. CrossRef - The importance of ongoing international surveillance for Creutzfeldt–Jakob disease
Neil Watson, Jean-Philippe Brandel, Alison Green, Peter Hermann, Anna Ladogana, Terri Lindsay, Janet Mackenzie, Maurizio Pocchiari, Colin Smith, Inga Zerr, Suvankar Pal
Nature Reviews Neurology.2021; 17(6): 362. CrossRef - Case series of Creutzfeldt-Jakob disease in a third-level hospital in Quito
Germaine Eleanor Torres Herrán, Andrés Damián Ortega Herrera, Braulio Martinez Burbano, Marcos Serrano-Dueñas, María Angélica Ortiz Yepez, Raúl Alberto Barrera Madera, Luis Alfredo Masabanda Campaña, Guillermo David Baño Jiménez, Denny Maritza Santos Salt
BMC Neurology.2018;[Epub] CrossRef - An Evaluation of Rapidly Progressive Dementia Culminating in a Diagnosis of Creutzfeldt–Jakob Disease
Parmvir Parmar, Curtis L. Cooper, Daniel Kobewka
Case Reports in Infectious Diseases.2018; 2018: 1. CrossRef - Diffusion-weighted MRI abnormalities antedate the onset of sporadic Creutzfeldt-Jakob disease
Keisuke Suzuki, Akiko Kawasaki, Takahide Nagashima, Koichi Hirata
Neurology.2016; 87(8): 843. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite