Basal Ganglia Syndrome in a Male With an XK Gene Variant but Without XK Disease (McLeod Syndrome)
Article information

Dear Editor,
XK disease (previously called McLeod syndrome) is a rare X-linked disorder caused by mutations in XK. A defining feature of this disease is an abnormal red blood cell (RBC) antigen profile, with no expression of Kx and decreased expression of Kell antigens (the “McLeod phenotype”) [1]. This clinical syndrome comprises a spectrum of systemic and neuropsychiatric manifestations. Chorea develops in 90% of patients [2]. Patients with the RBC McLeod phenotype but minimal or no neurological features have been identified [3]. XK functions as a scramblase and partners with VPS13A, which is responsible for VPS13A disease (formerly called “chorea-acanthocytosis”) [4,5].
We recently evaluated a patient with chorea and cognitive decline who carried a missense variant of XK; however, his RBCs lacked the McLeod antigen expression profile, which is required to make a diagnosis of XK disease [6]. In addition, he lacked other typical features of XK disease, such as creatine kinase (CK) and liver enzyme elevation, cardiomyopathy, peripheral neuropathy, and myopathy [6].
A 67-year-old man of Scottish/German ancestry presented with an 11-year history of gradually progressive chorea that was most prominent in his arms. He had slow horizontal and vertical saccadic eye movements with interruptions in smooth pursuit. Other neurological features, including reflexes, were normal. Cognitive screening revealed impairment in the working memory and executive function domains. Six months later, chorea was equally prominent in both lower extremities, with involvement of the trunk and minimal facial movements. He had an unsteady and lurching gait. Formal psychiatric evaluation revealed significant depression and alcohol abuse. He was emotionally labile and mildly impulsive.
At age 70, he complained of dysphagia with liquids, which was confirmed by modified barium swallow testing. Formal cognitive test results were unchanged, although he reported more difficulties with short-term memory, word-finding difficulties, and an inability to complete his thoughts at times. Chorea had progressed despite treatment with 24 mg of deutetrabenazine twice daily and 7.5 mg of aripiprazole once daily. He experienced frequent falls, one with syncope. Cardiac workup via electrocardiogram (ECG) and echocardiography was unremarkable. At age 73, his chorea was unchanged, but he experienced frequent falls. On examination, he had chorea affecting the trunk and all extremities (upper > lower), which increased while talking. His Montreal Cognitive Assessment (MoCA) score was 8/30 with prominent frontal executive dysfunction, difficulty with visuospatial tests and memory impairment. He was unable to perform the three-part Luria test.
The patient was one of eight siblings. One sister developed parkinsonism at age 66, followed by progressive language difficulties, memory impairment, right-sided apraxia, and right foot dystonia. Levodopa (300 mg/day) was discontinued due to severe drowsiness. She was wheelchair-bound after four years. Cranial magnetic resonance imaging (MRI) showed mild generalized volume loss with symmetric prominent hypointensities in the corpus striatum, substantia nigra and dentate nuclei but sparing of the thalami (Figure 1B). Her clinical profile was most consistent with corticobasal syndrome. She died at 76 years of age without undergoing XK gene testing or postmortem examination.
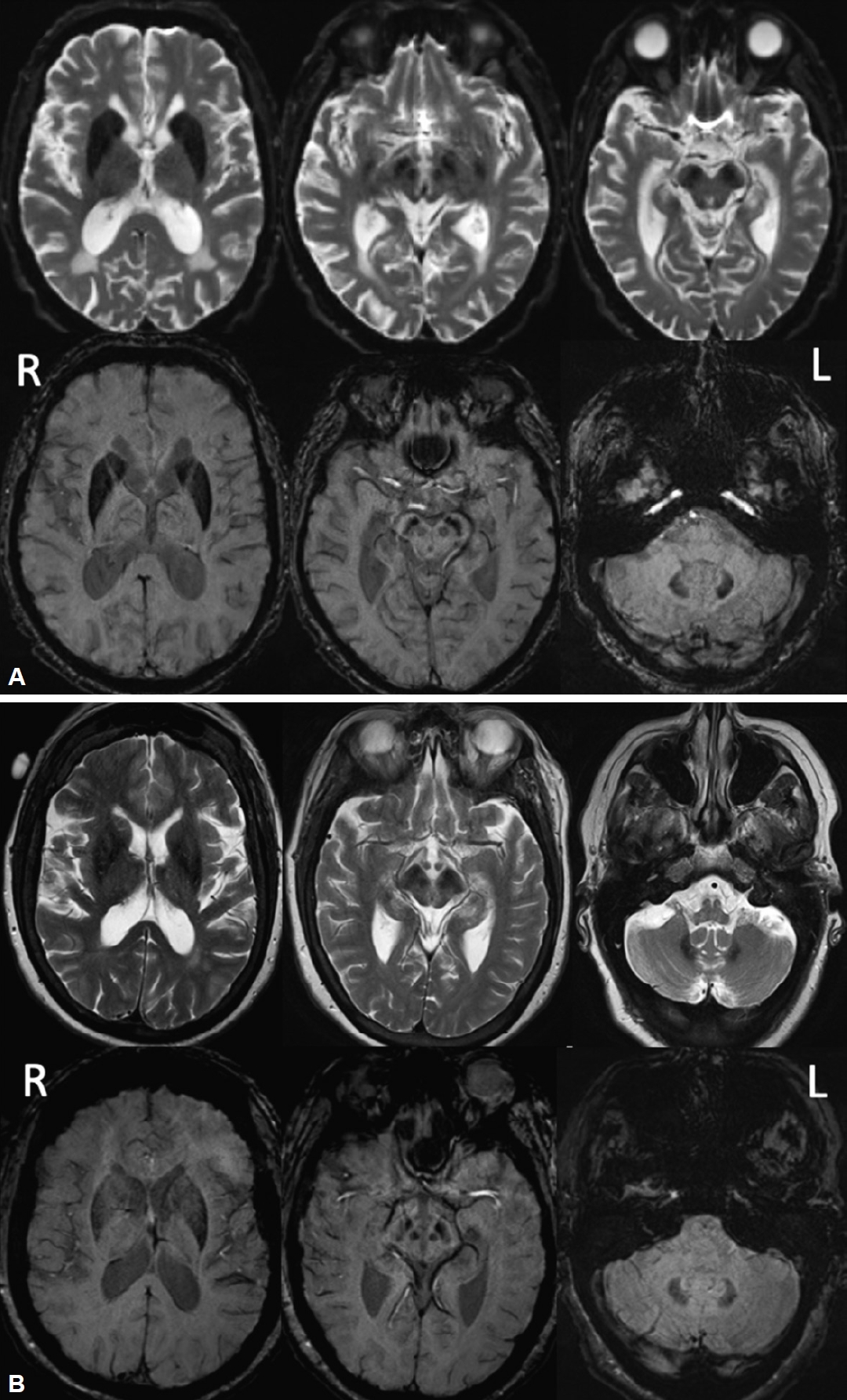
Cranial MRI of our patient and his sister. A: FLAIR images (top row) and SWI (bottom row) of our patient. Symmetric prominent hypointensities of the corpus striatum, substantia nigra and dentate nuclei with sparing of the thalamus were most evident on the susceptibility weighted and T2/FLAIR images. Cranial CT was normal without evidence of calcification. B: FLAIR images (top row) and SWI (bottom row) of his sister with the CBS phenotype. Mild generalized volume loss with symmetric hypointensities in the corpus striatum, substantia nigra and dentate nuclei with sparing of the thalamus were observed. MRI, magnetic resonance imaging; FLAIR, fluid attenuated inversion recovery; SWI, susceptibility weighted imaging; CT, computed tomography; R, right; L, left.
Another sister presented with prominent memory problems. One brother suffered from alcoholism, and another was diagnosed with bipolar disorder and committed suicide at age 49. On autopsy, no specific brain abnormalities were reported. Neither parent had neurologic symptoms according to the patient. His father died from pneumonia, and his mother died from cardiac disease. Family history showed that all individuals in the succeeding generation were asymptomatic.
The patient’s routine laboratory test results, including liver enzymes, CK, and repeated peripheral blood smears, were normal. His serum ferritin concentration was mildly elevated at 832 ng/mL (reference: 30.3–565.7 ng/mL). ECG, echocardiography, abdominal computed tomography (CT), nerve conduction studies, and electromyography results were normal.
Cranial MRI showed prominent symmetric hypointensities of the striatum, substantia nigra and dentate nuclei with sparing of the thalamus that were most evident on the susceptibility weighted and T2/fluid attenuated inversion recovery images (Figure 1A), suggestive of iron accumulation/deposition. A head CT did not show abnormal calcium deposition. There was no disproportionate caudate atrophy.
Kell blood group system serologic typing of RBCs revealed that K+k+, Kp(a-b+), Jsb+, and Kx+ levels were comparable to those of the controls and thus not consistent with the McLeod phenotype [6]. Sequencing of the XK gene revealed a novel missense variant, c.121T>G, in exon 1 causing a p.Leu41Val amino acid change that is predicted to be located in the 2nd transmembrane-spanning region of the XK protein [1,6]. This variant is classified as PM2 (moderate evidence of pathogenicity) according to ACMG guidelines. Chorein Western blotting analysis demonstrated normal levels of chorein (VPS13A).
The results of testing for the following trinucleotide repeat disorders were unremarkable: Huntington disease (19/21 repeats), Huntington disease-like 2 (14/15 repeats), SCA17 (37/38 repeats), and C9orf72 (2/2 repeats). No significant variants were found in genes related to iron dysmetabolism, including ATP13A, COASY, CP, C9orf12, FA2H, FTL, PANK2, PLA2G6, and WDR45, or in other genes potentially related to movement disorders, including AP4M1, CRAT, DCAF17, FUCA1, GJA1, GTPBP2, KIF1A, REPS1, SCP2, SLC25A42, SQSTM1, and VPS13A. Whole-genome sequencing (WGS) revealed only the previously identified XK gene variant.
Here, we describe the clinical profile of a patient who had a missense variant in the XK gene and normal expression of Kell and Kx red cell antigens. Other likely causes of his neurological presentation, including genetic disorders associated with iron deposition, were excluded. Acanthocytes were not detected; however, they can be absent in patients with XK disease [7].
We did not observe any typical clinical findings of XK disease, apart from progressive chorea and dementia; however, WGS detected only the XK gene variant and no other abnormalities. Although we were unable to diagnose XK disease in this patient for these reasons, extensive metabolic and genetic evaluations were also unremarkable. Although proving causality is beyond the scope of this article, we hypothesize that the XK variant results in impaired basal ganglia functioning but does not interfere with the expression of Kell and XK proteins on the erythrocyte membrane. This may be because only XK is translated in the brain and other neuronal tissues and is independent of Kell protein expression [1].
We acknowledge that these findings are the obverse of previously reported cases in which patients were found to have the erythrocyte McLeod phenotype but no or minimal neurological features [3]. These apparently complementary results may lead to further insights into the function of the XK scramblase as well as the mechanisms of lipid membrane trafficking in various organ systems.
Notes
Ethics Statement
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1975 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from the index patient.
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
Western blotting analysis for chorein was performed by G. Kwiatkowski and Dr. Benedikt Bader with the financial support of the Advocacy for Neuroacanthocytosis Patients and of the ERA-net E-Rare consortium EMINA (European Multidisciplinary Initiative on Neuroacanthocytosis; BMBF 01GM1003) in the labs of Profs. Hans Kretzschmar (Neuropathology) and Adrian Danek (Neurology) at Ludwig-Maximilians-Universität Munich, Germany. This study did not receive additional funding.
Author contributions
Conceptualization: Jeryl Ritzi T. Yu, Ruth H. Walker, Ilia Itin. Formal analysis: Adrian Danek, Connie M. Westhoff, Sunitha Vege. Project administration: Jeryl Ritzi T. Yu, Ruth H. Walker, Ilia Itin. Writing—original draft: Jeryl Ritzi T. Yu. Writing—review & editing: all authors.