Leucine-Rich Repeat Kinase 2-Linked Parkinson’s Disease: Clinical and Molecular Findings
Article information
Abstract
Mutations in Leucine-rich repeat kinase 2 (LRRK2) gene are the most common cause of sporadic and familial late onset Parkinson’s disease (PD). The G2019S common mutation has been identified about 1% of sporadic cases and 4–7% of familial cases. Over 50 variants have since been identified in LRRK2, and at least 7 of these are confirmed to be pathogenic. In addition to pathogenic mutations, several common polymorphisms in the LRRK2 gene (G2385R and R1628P) have been identified that may explain up to 10% of sporadic PD in Asian populations. LRRK2 is a large complex multidomain protein with 2,527-amino-acid and the molecular weight is 286 kDa. LRRK2 multidomain protein consists of a catalytic core domain, kinase domain and a number of putative protein-protein interaction domains. LRRK2 mutations found in PD families, including the G2019S and I2020T mutations show increased intrinsic kinase activity, when assessed with myelin basic protein as substrate. The modification of LRRK2 GTPase and kinase activity affecting residues in the ROC, COR and mitogen-activated protein kinase kinase kinases domains is believed to lead to neuronal cell death, but the pathways involved remain unclear. A number of in vivo models in C. elegans, D. melanogaster and mice have been developed to study the patho/physiological function of LRRK2. Based on current literature, a toxic gain of function in LRRK2 kinase activity is a possible pathophysiologic mechanism and thus inhibition of kinase activity in experimental models offers a potential therapeutic strategy for LRRK2-linked PD.
Parkinson’s disease (PD) is a devastating neurodegenerative disorder which affects 1–3% of the population over the age of 65. It is characterised by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc). Symptoms include resting tremor, postural instability, and slowing of physical movement (bradykinesia). Patients may also suffer from non-motor symptoms including dementia, mood disorders, hallucinations and sleep difficulties. Besides the sporadic form of PD, there are familial forms which are caused by single gene defects. One of these genes is Leucine-rich repeat kinase 2 (LRRK2, also referred to as PARK8), which is the most common cause of genetic form of PD, accounting for around 5–7% of all familial cases worldwide. Mutations in the LRRK2 gene that lead to PD are most common in the people from North African, Basque, Portuguese and Ashkenazi Jewish descent, but occur in almost all ethnic groups.
LRRK2 is a large and a complex multidomain protein that codes for protein rich in amino acid leucine and it functions as a kinase, acts as a mediator in synaptic endocytosis, regulation of neuronal survival, apoptosis, maintenance of neuritis and protein-protein interactions like signal transduction and in assembly of cytoskeleton and possibly functions as a scaffolding protein for cell signalling pathways. Thus LRRK2 might be an important physiological protein with a central role to play in a variety of neurodegenerative diseases.1,2
Genetics and Clinical Studies of LRRK2
LRRK2 was originally identified as a novel locus on chromosome 12p11.2-q13.1 that co-segregates with autosomal dominant Parkinsonism in a large Japanese family from Sagamihara region in Japan.3 In 2004, LRRK2 mutations (Y1699C and R1441C) were identified in European families (German Canadian and Western Nebraskan) 4 and Y1654C and R1396G mutations in British and Spanish families. Some authors named the gene product dardarin from the Basque word dardara meaning tremor, as it was a common symptom.5 In 2005 the G2019S mutation of was identified as the most common genetic cause of PD in Europeans, North African Berbers and Ashkenazi Jewish populations. 6–8 This G2019S common mutation has been identified about 1% of sporadic cases and 4–7% of familial cases.9 Over 50 variants have since been identified in LRRK2, and at least 7 of these are confirmed to be pathogenic. Pathogenic mutations are concentrated in the catalytic center of the protein, including R1441C/G/H in the renin angiotensin system of complex protein (ROC) domain, Y1699C in the C-terminus of ROC (COR) region, and G2019S and I2020T in the kinase domain.10 In addition to pathogenic mutations, several common polymorphisms in the LRRK2 gene (G2385R and R1628P) have been identified that may explain up to 10% of sporadic PD in Asian populations.11–13 Muta tions in LRRK2 gene are the most common cause of sporadic and familial late onset PD.
LRRK2 and Its Multi Domain Structure and Its Cellular Localization
LRRK2 is a large complex multidomain protein with 2,527-amino-acid and the molecular weight is 286 kDa. LRRK2 multidomain protein consists of a catalytic core domain, kinase domain and a number of putative protein-protein interaction domains (Figure 1). The catalytic core domain consists of a ROC domain, which belongs to the Ras GTPase superfamily, followed by a COR domain immediately before the kinase domain. The kinase domain of LRRK2 shows similarity to mitogen-activated protein kinase kinase kinases (MAPKKK) belongs to a serine/theronine and tyrosine kinase superfamily, which plays a central role in mediating cellular stress events. The protein-protein interaction domains include N-terminal armadillo (ARM) domain, ankyrin (ANK) repeats, 13 leucine-rich repeat regions (LRR), and 7 C-terminal WD40 repeats.14 LRR containing proteins participate in many biologically important processes, such as hormone-receptor interactions, enzyme inhibition, regulation of gene expression, apoptosis, and regulation of cytoskeletal dynamics, cell adhesion cellular trafficking and neural development.15 WD40 is also a conserved protein-protein interaction domain, that involved in a wide range of cellular functions, including signal transduction, mRNA processing, transcription, cytoskeletal assembly and mitochondrial fission.
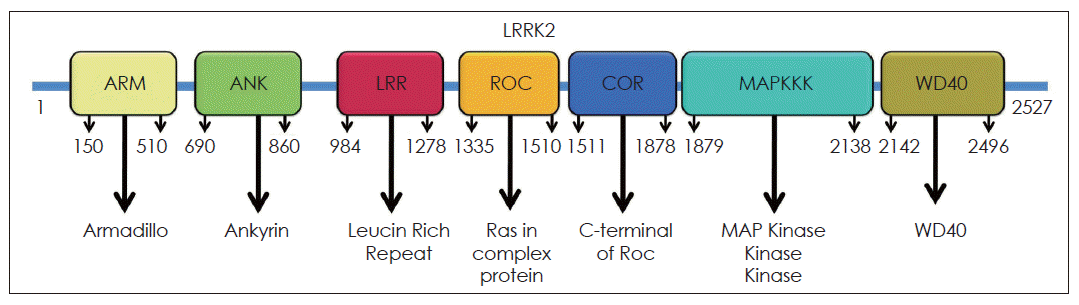
Structure of LRRK2. LRRK2 has 2527 amino acids and contains ARM, ANK, LRR, ROC, COR, MAPKKK and WD40 domains. LRRK2: Leucine-rich repeat kinase, ARM: armadillo, ANK: ankyrin, LRR: Leucine-rich repeat kinase, ROC: Renin-angiotensin system of complex protein, COR: C-terminus Of ROC, COR: C-terminus Of ROC, MAPKKK: mitogen-activated protein kinase kinase kinases.
LRRK2 is expressed throughout the human body with the highest levels of expression in the brain as well as in the peripheral organs including the heart, placenta, lung, kidney, skeletal muscle, pancreas and leucocytes. In the brain LRRK2 is expressed in the cortex, striatum, olfactory tubercle, thalamus, hippocampus, septal nuclei, and substantia nigra.16–18 For cellular localization, LRRK2 is predominantly present in the cytoplasm, and a portion of LRRK2 is associated with the membranes of organelles such as mitochondria, golgi apparatus, plasma membranes and endo plasmic reticulum, synaptic vesicles like lysosomes, endosomes, transport vesicles and lipid rafts and microtubules. A prominent role of LRRK2 association with membranous and membrane-bound organelles suggests that LRRK2 plays a role in the regulation of synaptic function is probably through vesicle synthesis and transport or through regulation of membranous structures or protein turnover and cytoskeletal organization. The presence of both enzymatic domains ROC and MAPKKK, as well as protein interaction domains, also indicates that LRRK2 might serve as a scaffold for multiprotein signaling complexes.
Functional Activity of LRRK2
LRRK2 has a dual enzyme activity known as kinase and GTPase activity that is flanked by LRR and WD40 domains. The kinase activity assists in the transfer of a phosphate group from the energy molecule ATP to amino acids in certain proteins. This phosphate transfer is called phosphorylation, and it is an essential step in turning on and off many cell activities. LRRK2 GTPase enzyme activity is associated with a region of the protein called the ROC domain. The ROC domain may act as a molecular switch that controls the overall shape of the LRRK2 protein.
Kinase activity of LRRK2
The modification of LRRK2 GTPase and kinase activity affecting residues in the ROC, COR and MAPKKK domains is believed to lead to neuronal cell death, but the pathways involved remain unclear. The combination of GTPase activity mediated via the ROC-COR tandem domain and kinase activity of the MAPKKK domain also suggests a complex role for LRRK2 in cell signalling.19 In vitro studies show a role of LRRK2 in pathogenicity by demonstrating that transiently transfected mutant LRRK2 causes neurodegeneration in SH-SY5Y neuroblastoma cells and in primary neurons. The mutation G2019S results in the gain of function causing increased kinase activity of LRRK2 leading to autosomal dominant PD. Several groups reported kinase activities of the mutants using in vitro kinase assays and found that only the G2019S mutant exhibited increased kinase activity, while the other mutants, R1441C, Y1699C and I2020T showed uncertain and sometimes conflicting results. These in vitro findings suggest that increased kinase activity is likely the pathogenic mechanism that could result in the dysregulation of vesicular trafficking and transport20–22 and alterations in neurite outgrowth.23,24 More convincing evidence for aberrant kinase activity of LRRK2 as a suspect in LRRK2-mediated neuropathogenesis comes from a series of in vitro experiments using cultured neurons where nearly all pathogenic mutations of LRRK2 are neurotoxic and inhibition of kinase activity can eliminate this toxicity.19,21,25,26 In addition, blocking of putative phosphorylation sites of LRRK2 kinase results in reduced neurotoxicity in fly models.27,28
GTPase activity of LRRK2
The ROC domain of LRRK2 shares homology with the Ras related GTPase superfamily, responsible for regulating diverse cellular processes, such as mitogenic signalling.14 The activation of the Ras related GTPase requires a conversion of the GDP bound state to the GTP bound conformation (Figure 2).29,30 The GTPase family functions as a molecular switch that requires guanine nucleotide exchange factor (GEF) and GTPase activating protein (GAP) activity to shuttle between its GTP bound active state and GDP bound inactive state. Therefore, proving whether the GTPase domain of LRRK2 is functional was difficult at first without knowing the identity of LRRK2’s specific GEF. Disruption of the critical residues of the GTPase domain mutants (K1347A and T1348N), impair GTP binding, significantly decline LRRK2 kinase activity whereas others reported that loss of LRRK2 binding to GTP, GDP or GMP had no effect on its kinase activity.28 In vitro GTP hydrolysis activity of LRRK2 has been repeatedly reported to be low down. However, LRRK2 purified from transgenic mice brain shows high levels of GTPase activity.29 These apparently inconsistent results might be explained by the lack of the specific GEFs or GAPs in the in vitro experiments. Mutations close to the GTPase domain such as R1441C and R1441G reduce GTP-hydrolysis activity and prolong the GTP-bound state that, in turn, enhances LRRK2 kinase activity.14,29,30 It is not yet clear if functional GTPase activity is required for kinase activity. In conclusion, the accumulated data suggest that LRRK2 contains functional GTPase and kinase activities. Of the PD specific mutants, G2019S enhances kinase activity and R1441C decreases GTPase activity. However, it remains to be clarified whether any mutants other than G2019S affect its kinase activity and whether GTPase activity is a prerequisite for kinase activity.14,29,30

GTPase activity of LRRK2. GTPases exist in a GDP bound (inactive) and GTP bound (active) form, which have different conformations. Conversion from the GDP to the GTP bound form is caused by nucleotide exchange, catalyzed by guanine nucleotide exchange factors. Conversion from the GTP to the GDP bound form occurs by intrinsic GTP hydrolysis, which is facilitated by GTPase activating proteins. GTP: GTPase activating protein.
LRRK2 and Its Phosphorylation Substrates
The identification and characterization of LRRK2 interactors and substrates of its kinase activity would provide the best way to understand the molecular pathogenesis of LRRK2 associated PD. Interestingly, most of the LRRK2 interacting proteins identified till date are cytoskeleton and trafficking proteins. LRRK2 functions as a serine/threonine kinase that can undergo autophosphorylation. In vitro, it can phosphorylate the generic kinase substrate myelin basic protein (MBP). Several other likely more physiological substrates such as moesin, ezrin and radixin have been identified. LRRK2 phosphorylates itself predominantly within its GTPase/ROC domain1. Phosphorylation of a threonine 1343 (T1343) was consistently observed with several different methods and confirm that this residue authentically resulted from autophosphorylation by using a kinase dead LRRK2 molecule. T1343 residue is predicted to be involved in GTP binding in either of the two models of the ROC domain that are available. Therefore, T1343 will impact GTPase function, which suggests an intriguing intermolecular regulation of the GTPase domain by the kinase domain. By using MBP as a test substrate, West et al. measured the activity of LRRK2 protein and determined that it possesses mixed lineage kinase (MLK) activity. LRRK2 protein has also been demonstrated to undergo autophosphorylation, where the intrinsic GTP binding activity modulates downstream kinase activity. Other regulatory sequences suggested to modulate the kinase activity of LRRK2, include the N and C-termini. The N-terminus of LRRK2 has been suggested to have an inhibitory effect and C-terminal tail is required for full kinase activity.21,31
Indeed, individual LRRK2 mutations found in PD families, including the G2019S and I2020T mutations, show increased intrinsic kinase activity, when assessed with MBP as substrate. 25 Hsu et al. proved that LRRK2 interacts with MKK6 in mammalian cells by overexpressing tagged LRRK2 and MKK6. The authors also have found that expression of MKK6 has increased in the presence of LRRK2 and vice versa. It is more intriguing that overexpression of these two proteins has changed the subcellular localization of MKK6. The mechanisms of increase at protein level and the physiological meaning of the change of subcellular localization still need to be further investigated. Furthermore, authors have used C. elegans to examine possible physiological function of the interaction between LRRK2 and MKK6. The result is interesting but need to be interpreted carefully. This biochemical interaction between LRRK2 and MKK6 and offered valuable information of LRRK2 might be related to the stress response.
LRRK2 and Its Signaling Pathways
Multiple LRRK2 PD mutants show enhanced toxicity, causing significantly greater cell death than the wild-type LRRK2 protein in cell lines and primary neurons. Over-expression of wild-type and mutant (G2019S) LRRK2 has been shown to reduce the neurite length and branching in primary neuronal cell cultures, whereas LRRK2 deficiency results in increased neurite length and branching.19 Neurite shortening has also been reported in differentiated SH-SY5Y transfected with G2019S mutant LRRK2, which was also shown to exhibit an increase in autophagic vacuoles.21,25,32 Thus, most PD mutations appear to cause cell death by altering some other feature of LRRK2 biology but that nonetheless requires intact kinase function. This proves that LRRK2 is a signaling molecule which regulates the function of other proteins and that kinase activity is one key part of the signaling pathways. Presumably, LRRK2 becomes pathogenic when the kinase is hyperactive or misregulated and this may involve signaling pathways.
There are two pathways responsible for cell survival that may involve products of genes associated with PD. These include MAPK (ERK1/2) signaling pathway and the AKT/GSK-3β dependent pathway. The LRRK2 gene encodes a MAPKKK protein. MAPKs downstream targets can be either in the cytoplasm or in the nucleus, when the substrates are factors or transcriptional co-regulators. The MAPK pathway mediates signal transduction from cell surface receptors to downstream transcription factors that lead to cellular responses such as cell proliferation, growth, motility, survival and apoptosis. The MAPKs regulated three different groups of pathways, ERK, p38MAPK and c-Jun N-terminal kinases (JNK). Potential cross-talk between LRRK2 and the ERK pathway was first suggested by the decrease in basal level of pERK in cells expressing LRRK2 wild-type, its mutant and their corresponding deletion mutants which were consistent to its predicted role as a protein scaffold facilitating the activation of the ERK1/2 pathway.33 One study demonstrates mediation of the ERK pathway in neurite shortening and autophagy in G2019S differentiated SH-SY5Y cells.32 This hypothesis was later confirmed in two cellular models (HEK293 and SH-SY5Y), whereby the authors demonstrated that wild-type LRRK2 protein is involved in the activation of the ERK pathway, and also confers protection in response to hydrogen peroxide mediated oxidative cellular stress.34 In this model, LRRK2 mutants were unable to appropriately activate the pathway, leading to cell death. This is because as a protein scaffold facilitating the activation of the ERK1/2 pathway, over-expressing LRRK2 wild type or mutants would inevitably suppress the activation of the ERK1/2 pathway which may come with detrimental consequences to cell growth and maintenance of cell viability.35
LRRK2 phosphorylates MKK4 and MKK7 within the activation loop where phosphorylation primes the kinase domain for activity, leading to downstream activation of JNK pathway, consistent with the assignment of LRRK2 as a potential MAPKKK. However, over-expression of LRRK2 protein in cells does not lead to an obvious up-regulation of phosphorylated JNK or c-Jun as might be anticipated, suggesting either a lack of necessary cofactors or that LRRK2 phosphorylation of MAPKK proteins does not occur with high efficiency in cells. An emerging theme in the MAPK pathway suggests that scaffolding proteins play critical roles in mediating phosphorylation events that are otherwise unlikely to occur.36 LRRK2 may serve as a protein scaffold for MAPK signaling, where identification of binding partners and necessary cofactors are required before definitive assignment of LRRK2 into the MAPK pathway. The potential role of MAPKs in the pathogenesis of PD is supported by additional experimental evidence including the activation of JNK in several PD animal models, as well as the neuroprotection conferred by upstream inhibition of the pathway.37
In vivo Studies of LRRK2
A number of in vivo models in C. elegans, D. melanogaster and mice have been developed to study the patho/physiological function of LRRK2. The three published D. melanogaster models provide conflicting information. One model over-expressing the G2019S mutant and a loss-of-function model of dLrrk, the LRRK2 paralog in D. melanogaster, report retinal degeneration, selective loss of dopaminergic neurons, motor impairment and reduced lifespam.35,38 In contrast, a loss-of-function model lacking the dLrrk kinase domain resulted in increased oxidative stress sensitivity to H2O2 with no signs of neurodegeneration. 35,39 C. elegans over-expression models point towards a role of LRRK2 in regulating stress response and neurite outgrowth40 whereas the paralog gene, lrk-1, seems to be involved in regulating synaptic vesicle proteins polarity in a knockout model. Recent phylogenetic analyses of the COR, Roc and kinase domains of LRRK2 demonstrate that LRRK2 emerged from a duplication that was prior to the acquisition of the specific LRRK2-repeats at the N-terminal of the protein. Paralogs of LRRK2 such as LRRK1 or protostome LRRK encoded proteins lack this repeat, suggesting that D. melanogaster or C. elegans may not be good models to understand human LRRK2 function.
Four studies of LRRK2 fly models have reported using D. melanogaster to investigate the in vivo functions of dLRRK. In 2007, Lee et al.38 shows that mutant flies lacking dLRRK exhibited impaired locomotive activity and a significant reduction of tyrosine hydroxylase (TH) immunostaining in dopaminergic neurons. The dopaminergic neurons are unaltered in numbers, but they display abnormal morphology, suggesting that they are under pathogenic stress or undergoing slow degeneration. Two other studies in 2008, Wang et al.39 and Imai et al.27 proved that TH deficits in mutant fly carrying deletion of dLRRK. Instead, they observed unchanged numbers of TH positive neurons in these mutants, indicating that dLRRK is dispensable for the survival of dopaminergic neurons. In addition, wang et al. showed that mutant flies containing C-terminal kinase domain truncated dLRRK are selectively sensitive to H2O2, but not to paraquat, rotenone or β-mercapto-ethanol. By contrast, Imai et al.27 showed that dLRRK null flies are relatively resistant to general oxidative stress, such as paraquat and H2O2 treatment, compared to wildtype flies. Although the exact role of dLRRK in oxidative stress remains unclear, all studies in fly models reported to date consistently demonstrate that dLRRK is not essential for the early development and viability of dopaminergic neurons. Liu et al.41 2008, found that treatment of L-DOPA improved the motor impairment of transgenic flies caused by LRRK-G2019S but not the degeneration of TH positive neurons. This showed that even over-expressing wild-type human LRRK2 led to the toxicity of dopaminergic neurons and impairment of motor function (although to a lesser degree than LRRK2 G2019S),41 whereas the other indicated that over-expressing wild-type dLRRK did not affect the number of dopaminergic neurons or motor function.
Unfortunately, none of the rodent models developed so far (knock in, knockout and transgenic for LRRK2) show any clear LRRK2-associated phenotype. Despite very limited in vivo information about LRRK2 function is available, strong evidence suggests a crucial role of LRRK2 kinase activity in the pathogenesis of the disease, probably by regulating cytoskeletal signaling pathways and neurite outgrowth. In 2007, Biskup et al. first to report the generation of LRRK2 KO (knock out), it is also not surprising that no proper result was proved. Although without showing any experimental data, a study by Wang et al.42 indicated that LRRK2 KO mice survive normally, and that they do not develop any obvious neuropathological abnormalities or motor dysfunctions. Indeed, no loss of dopaminergic neurons or motor behavioral deficits was observed even at 24 months of age in LRRK2 KO mice. This result, along with the study showing the developmental expression levels of LRRK2, suggests that the role of LRRK2 in early embryonic development is negligible, but may be important for cellular function at the adult stage.42
In the study of LRRK2 transgenic mice, two mouse models with the normal or mutant LRRK2 using an advanced form of genetic engineering called bacterial artificial chromosome genetics (BAC). Two laboratories generated BAC-transgenic mice expressing murine FLAG-tagged LRRK2 and human LRRK2,18 whereas the third indicated the usage of a tetracyline-regulated system for the transgenic expression of human G2019S LRRK2.42 The BAC transgenic approach has been successfully used in establishing mouse models for neurode-generative diseases and is expected to contribute to an understanding of the disease mechanisms in vivo. Melrose et al.18 previously reported the generation of BAC transgenic mice producing human LRRK2 wild type or mutants. Although no information was given about the viability of TH positive neurons, it was indicated that BAC transgenic mice over-expressing LRRK2 wild type, mutant G2019S or Y1699C did not show an overt behavioral phenotype. Consistent with these observations, tetracycline-regulated transgenic mice producing LRRK2 G2019S were also reported to be spared of obvious neuropathologies or motor abnormalities.42 Interestingly, a more recent study by Li et al.43 suggests that BAC transgenic mice expressing the human LRRK2 R1441C mutant develop typical motor function deficit related to PD. These mice are associated with the degeneration of TH positive axons and tauopathy, as well as TH positive cell atrophy, despite lacking obvious loss of midbrain TH positive cells.
Conclusion
LRRK2 is the most common cause of sporadic and family forms of late onset PD worldwide. The challenge is to further unravel its biological functions in vivo models of LRRK2. Based on current literature, a toxic gain of function in LRRK2 kinase activity is a possible pathophysiologic mechanism and thus inhibition of kinase activity in experimental models is rigorously examined by many investigators with the aim to develop a therapeutic strategy for PD.31 The identification of drug targets will eventually lead to clinical trials and hopefully these could translate into better therapies for patients suffering from PD.
Notes
The authors have no financial conflicts of interest.