Clinical and Imaging Profile of Patients with Joubert Syndrome
Article information

Abstract
Objective
Joubert syndrome (JS) is a rare syndrome characterized by ataxia and the molar tooth sign (MTS) on imaging. The present study aims to explore the clinical and radiological features in a cohort of patients with JS.
Methods
This was a retrospective chart review of patients with JS evaluated by movement disorder specialists.
Results
Nine patients were included in the study. All patients had facial dysmorphism and ocular abnormalities, and 4 patients had dystonia. Ocular tilt reaction and alternate skew deviation (66%) were the most common ocular abnormalities. Horizontally aligned superior cerebellar peduncles were observed in all four patients with diffusion tensor imaging, with a lack of decussation in three. Exome sequencing performed in four patients revealed novel variants in the MKS1, CPLANE1, and PIBF1 genes.
Conclusion
Facial dysmorphism, ocular abnormalities and classical imaging findings were observed in all patients with JS. Apart from ataxia, dystonia and myoclonus are other movement disorders observed in JS.
Joubert syndrome (JS) is a ciliopathy with abnormalities in the structure and function of cilia in the brain, retinal photoreceptors, bile duct epithelium and microtubules of the kidney, resulting in multiorgan involvement [1,2]. JS is diagnosed by the presence of three cardinal features: hypotonia in infancy, molar tooth sign (MTS) on brain imaging, and developmental delay [1,3,4], with ataxia typically occurring later in the course of illness. Additionally, abnormal breathing patterns, abnormal eye movements, and facial dysmorphism can be present [1]. Movement disorders other than ataxia are rarely described [5-7]. The diagnosis of JS is highly dependent upon imaging, which shows classical MTS [8]. Genetic testing is also beneficial, with more than 30 causative genes identified [9]. The present study aims to characterize the spectrum of neurological involvement with a focus on movement disorders and other systemic involvement, blood investigations, and imaging abnormalities.
MATERIALS & METHODS
This is a retrospective chart review of patients with JS evaluated by movement disorder specialists at National Institute of Mental Health and Neurosciences, India from 2014 to 2019. Patients with JS diagnosed based on the presence of MTS on imaging in the background of clinical features consistent with JS, such as developmental delay, apneic episodes and/or ataxia, were included in the study. The available demographics, clinical history, examination details, and investigations, including imaging and genetic data, were documented. In addition, patient videos were also reviewed. The data were expressed using descriptive statistics. The Institute Ethics Committee at the National Institute of Mental Health and Neurosciences granted an ethical clearance waiver owing to the retrospective nature of the study with de-identified data being extracted from files, and informed written consents were obtained for publication of recorded videos (No: NIMH/DO/DEAN [Basic Science]/2020-21).
RESULTS
Nine patients (5 females, 55.5%) belonging to 8 families with a mean age of presentation of 10.3 ± 6.82 years (median: 12.5 years, range 10 months–24 years) were included. Of them, one patient (patient 3) was included in a previous report [7]. The demographic and clinical details are provided in Supplementary Table 1 (in the online-only Data Supplement).
Clinical features
Respiratory abnormalities such as apneic and hyperpneic episodes were seen in six patients in the neonatal period (Supplementary Table 1, Video 1 in the online-only Data Supplement). Developmental delay was noted in all patients, with severe developmental delay in 4 patients. One patient (patient 3) had generalized tonic clonic seizures (GTCSs) that were under control. All patients had facial dysmorphism, of which broad nasal bridges (n = 5) and low-set ears (n = 5) were most common (Supplementary Table 1 in the online-only Data Supplement, Figure 1). Head tilt was observed in 5 patients, and arachnodactyly and kyphosis were observed in 2 patients.
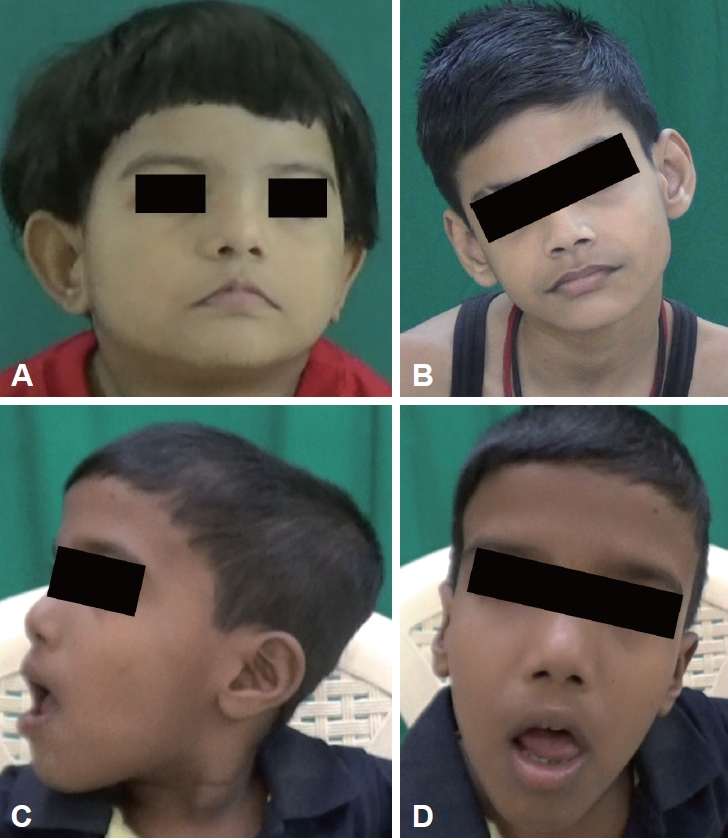
Facial dysmorphisms seen in Joubert syndrome. Broad nasal bridge (A), frontal bossing (C, D), trapezoid mouth (A, D), everted lower lip (C, D), dolichocephaly (C), elongated face (B, D), and head tilt (B, D). A: Patient 1. B: Patient 6. C, D: Patient 7.
Neurological examination
Intellectual disability was noted in all patients, ranging from mild to moderate, with an intelligence quotient of less than 50 in 6 patients, while data were not available for the other 3 patients. Ocular abnormalities were seen in all patients (Supplementary Table 1 in the online-only Data Supplement), with congenital oculomotor apraxia (COMA) in 4 patients (Supplementary Video 2 in the online-only Data Supplement), ocular tilt reaction (OTR) in 6 patients (Supplementary Video 3 in the online-only Data Supplement) and alternate skew deviation (ASD) in 6 patients (Supplementary Video 4 in the online-only Data Supplement). Six patients had speech abnormalities, with hypernasality observed in 4 of these patients (Supplementary Video 5 in the online-only Data Supplement).
Movement disorders
A total of 4 patients had dystonia, of whom 2 patients had cervical dystonia (patients 4 and 9) (Supplementary Table 1 in the online-only Data Supplement), and 1 each had segmental dystonia involving the neck, trunk and upper limb (patient 6) (Supplementary Video 5 in the online-only Data Supplement) and generalized dystonia (patient 5). Multifocal myoclonus probably of reticular origin involving axial and proximal appendicular muscles present at rest and triggered by sound and action was noted in one patient who also had GTCS (patient 3). Four patients did not attain the gross motor milestone of walking, and gait ataxia was present in the other 5 patients.
Investigations
Imaging
All patients had MTS, deep interpeduncular fossa, vermian hypoplasia, superior cerebellar peduncle (SCP) thickening, fourth ventricular enlargement and batwing appearance of the fourth ventricle (Supplementary Table 2 in the online-only Data Supplement, Figure 2). Supratentorial abnormalities were observed in 3 patients (Figure 2). Diffusion tensor imaging (DTI) data were available for 4 patients, where horizontally oriented fibers were seen in all 4 and a lack of decussation was observed in 3 patients.
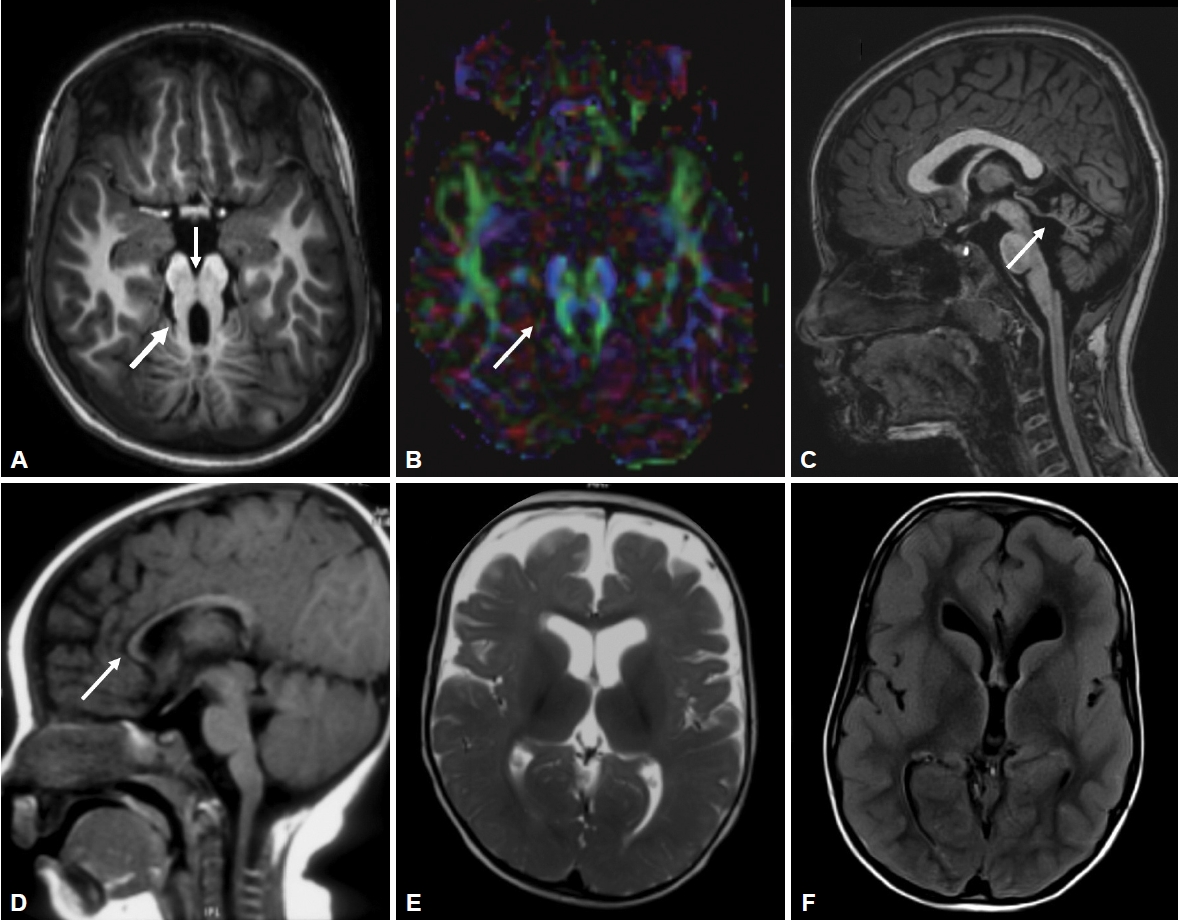
Magnetic resonance imaging findings in Joubert syndrome. (A) Molar tooth sign (thick arrow) and deep interpeduncular fossa (thin arrow). (B) Color coded fractional anisotropy map showing horizontally oriented superior cerebellar peduncles (thin arrow) and the absence of normal decussating horizontal fibers, (C) superior cerebellar (thin arrow) and vermian hypoplasia, (D) callosal atrophy (thin arrow), (E) frontal cerebral atrophy and (F) diffuse pachygyria. A-C: Patient 4. D: Patient 1. E: Patient 2. F: Patient 7.
Genetics
Clinical exome sequencing was performed in 4 patients, and abnormalities were observed in CPLANE1 in patient 3, MKS1 in patients 5 and 6, and PIBF1 in patient 9. Five variants were identified in total, and all were novel (Supplementary Table 2 in the online-only Data Supplement). All were either pathogenic or likely pathogenic variants except for a variant of unknown significance (chr5:g.37247731A>G;p. Trp24Arg) found in patient 3. As the variant was in the compound heterozygous trans configuration with a pathogenic variant and in a patient with a typical phenotype, the variant was deduced as a disease-producing variation.
DISCUSSION
JS is characterized by the classical features of hypotonia, apneic episodes in the newborn period followed by developmental delay, facial dysmorphism, ocular movement abnormalities and gradual development of ataxia [9]. The later mean age at presentation in our cohort compared to previous studies [10] might be due to referral bias. Additionally, in all our patients, apneic episodes disappeared by infancy, while the presence of prolonged apneic episodes is predictive of poor survival in JS [11]. Furthermore, none of our patients had multiorgan involvement, such as renal or cardiac issues.
Facial dysmorphism is common, with features including a long face, prominent forehead, epicanthal folds, ptosis, arched eyebrows, broad nasal bridge, prognathism, everted lower lip, trapezoid mouth, and tongue protrusion [12]. Craniofacial abnormalities have been suggested to be associated with ciliary dysfunction [13]. However, they are variable and nonspecific and can overlap with other hindbrain syndromes, and genetic heterogeneity may add to the variability [12].
Ocular abnormalities are one of the cardinal features of JS and include COMA, OTR, ASD, strabismus, nystagmus, ptosis, coloboma and retinopathy. COMA results from an inability to generate voluntary saccades compensated by head thrusts or frequent eye blinking. This irregularity in conjugate eye movements is a hallmark feature of JS. There can be frequent and periodic cyclic deviations of the eye, such as alternate gaze deviation, ASD, alternate torsional deviation, and wheel rolling torsional eye movements. Cyclic alternate torsional deviations, a type of OTR, are diagnostic of JS when seen in association with COMA [14,15]. It is characterized by spontaneous skew deviation, cyclotorsion of both eyes and paroxysmal head tilting, and it shares similarity with episodic apnea hyperpnea. These oculomotor abnormalities arise due to defects in cerebellar outflow pathways, nondecussation of the brainstem pathway and the loss of crossed inputs [15].
Movement disorders apart from ataxia are rarely described in patients with JS. Ataxia in JS occurs due to vermian hypoplasia and nondecussation of SCP [10]. Cervical dystonia has been previously reported in a few case reports [5,6]. In our cohort, 5 patients had head tilt, which could be due to either cervical dystonia or oculomotor abnormalities. Even though the presence of additional appendicular dystonia in 2 patients may favor cervical dystonia, the absence of typical dystonic neck spasm or head tremor may suggest oculomotor abnormalities as the cause for head tilt. Myoclonus may also rarely occur and has been previously reported by our group [7].
All patients with JS have cardinal magnetic resonance imaging features of MTS that occur due to deepened interpeduncular fossa, hypoplasia of the cerebellar vermis and thickened SCPs. Additionally, supratentorial findings can be associated, such as hippocampal malrotation, migration disorders, hydrocephalus, callosal dysgenesis and encephalocele (Figure 2). DTI in JS shows horizontal orientation of SCPs compared to vertically oriented SCPs in healthy subjects. The normal transverse decussation of SCPs at the level of inferior colliculi is visualized as a red dot on fractional anisotropy maps, and this is conspicuously absent in JS [16]. Of the 4 patients who had undergone DTI, all had horizontally oriented SCPs, while 3 had the absence of SCP decussation (Figure 2).
JS is a genetically heterogeneous disorder with more than 30 genes identified thus far that are involved in producing proteins important for ciliary function [9]. There is variability in the genotype-phenotype correlations, and certain clinical features, such as orofacial digital abnormalities, renal abnormalities, retinal dystrophy, and hepatic abnormalities, may be attributable to a specific genetic mutation [17]. Patient 3 had abnormalities in the CPLANE1 gene with pure JS except for GTCS and multifocal myoclonus. Orofacial digital abnormalities have been described in JS with CPLANE1 mutations. However, up to two-thirds of cases can have a pure JS phenotype [18]. Mutations in the MKS1 gene may present as a pure JS phenotype or can be associated with retinal dystrophy, retinal coloboma, and renal and hepatic involvement [15,19]. Patient 6 with MKS1 gene abnormalities had retinal coloboma. Patient 9 with a pure JS phenotype had abnormalities in the PIBF1 gene, which can present either as pure JS or with retinal dystrophy, cystic kidney disease, liver fibrosis, and polydactyly [20]. These three genes play a role in ciliary function, explaining their involvement in JS.
There are a few limitations to this study owing to the retrospective and cross-sectional nature. Systemic involvement in JS may develop over the course of the illness, and the absence of these symptoms at the time of evaluation may not imply the complete absence of these symptoms in the patient. Longitudinal studies are necessary to understand the temporal evaluation of symptoms in JS. A few patients seen at a very young age may not have the full spectrum of manifestations.
To conclude, Joubert syndrome is a rare neurological disorder with a wide range of phenotypic features and significant genotypic variability. Facial dysmorphism and ocular abnormalities aid in suspecting Joubert syndrome, and apart from ataxia, the movement disorder spectrum of Joubert syndrome may include dystonia and myoclonus.
Supplementary Material
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.21066.
Video 1.
An infant with JS (patient 2) showing apneic episodes along with cyclic alternating dysconjugate horizontal gaze deviation. Additionally, note the frontal bossing, low-set ears, broad nasal bridge, open trapezoid mouth and tongue protrusion.
Video 2.
Oculomotor apraxia with compensatory head thrust in patient 3.
Video 3.
Cyclic alternate torsional deviation with head tilt (a form of ocular tilt reaction) in patient 6.
Video 4.
Alternating hypertropic skew deviation of the right eye followed by the left eye in patient 5.
Video 5.
Hypernasality of speech. Cervical, truncal, and upper limb dystonia and ataxic gait were observed in patient 6.
Supplementary Table 1.
Clinical features of patients with Joubert syndrome
Supplementary Table 2.
Imaging and genetics of patients with Joubert syndrome
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
None.
Author Contributions
Conceptualization: Bharath Kumar Surisetti, Vikram Venkappayya Holla, Shweta Prasad, Pramod Kumar Pal. Data curation: Bharath Kumar Surisetti, Vikram Venkappayya Holla, Shweta Prasad, Koti Neeraja. Formal analysis: Bharath Kumar Surisetti, Vikram Venkappayya Holla, Shweta Prasad. Investigation: Bharath Kumar Surisetti, Vikram Venkappayya Holla, Koti Neeraja. Methodology: Bharath Kumar Surisetti, Vikram Venkappayya Holla, Pramod Kumar Pal. Project administration: Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Resources: Vikram Venkappayya Holla, Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Supervision: Vikram Venkappayya Holla, Ravi Yadav, Pramod Kumar Pal. Visualization: Vikram Venkappayya Holla, Pramod Kumar Pal. Writing—original draft: Bharath Kumar Surisetti. Writing—review & editing: Vikram Venkappayya Holla, Shweta Prasad, Koti Neeraja, Nitish Kamble, Ravi Yadav, Pramod Kumar Pal.