Dystonia Responsive to Dopamine: POLG Mutations Should Be Considered If Sensory Neuropathy Is Present
Article information

Abstract
The POLG gene encodes mitochondrial DNA polymerase, and mutations in this gene cause a spectrum of disorders related to mitochondrial DNA depletion or deletion. Dystonia has only rarely been reported as an early and prominent manifestation of POLG mutations. We report a case of a 30-year-old male presenting with lower limb dystonia with peripheral neuropathy and demonstrate that the dystonia was levodopa responsive (with video findings). Whole-genome sequencing revealed biallelic variants in the POLG gene: a known pathogenic variant [NM_001126131.2:c.2209G>C (p.Gly737Arg)] and a novel likely pathogenic variant [NM_001126131.2:c.3305A>C (p.Gln1102Pro)]. A genetic diagnosis was made before the appearance of more readily recognizable features of mitochondrial disease, allowing us to avoid invasive tissue biopsies or potentially deleterious treatments, such as sodium valproate. A POLG-related disorder should be suspected in cases of dystonia with peripheral neuropathy, and this diagnosis may have implications for further investigations and management.
The POLG gene encodes the catalytic subunit of mitochondrial DNA polymerase called DNA polymerase gamma. POLG is a nuclear gene responsible for mitochondrial replication. Mutations in this gene can result in quantitative mitochondrial depletion, typically manifesting as one of a heterogeneous group of recessive early onset syndromes or syndromes caused by qualitative mitochondrial deletions that often arise later in life [1].
POLG mutations represent the most common cause of inherited nuclear DNA-encoded mitochondrial disorders [1]. POLG-related disorders encompass a spectrum of disorders, including mitochondrial DNA depletion syndrome type 4A (Alpers) and 4B (mitochondrial neurogastrointestinal encephalomyopathy), mitochondrial recessive ataxia syndromes (sensory ataxic neuropathy, dysarthria, ophthalmoparesis, and spinocerebellar ataxia with epilepsy) and progressive external ophthalmoplegia type 1 (dominant and recessive). Pediatric patients have a predominance of seizures, hepatopathy, and lactic acidaemia, whereas patients developing symptoms in adulthood are more frequently affected by myopathy, sensory ataxia, and chronic progressive external ophthalmoplegia [2]. POLG mutations are also known to be implicated in one of many mitochondrial diseases associated with disorders of movement [3,4].
There are few reports in the literature of a POLG-related disorder with prominent dystonia at presentation [5]. We report a proband presenting with dystonia and peripheral neuropathy who was found to have biallelic disease-causing variants in the POLG gene.
CASE REPORT
A 30-year-old male born of nonconsanguineous parents presented with asymmetric tremor and dystonia, bradykinesia and sensory symptoms that began at 23 years of age. He first noted a tremor in his right leg, which was initially present on exertion only. In the following year, he noted some impairment in balance and coordination that was exacerbated with eye closure and in the dark. Subsequently, his leg tremor became more pronounced, spreading to involve the left leg. This tremor was associated with involuntary inversion of the feet and right thumb flexion, head posturing, and paresthesia in the right foot. Dystonia and tremor were absent when supine, and there was a diurnal component occurring only several hours after awakening. There was no relevant past medical history or family history.
On presentation, the patient had a weight of 73 kg and a height of 170 cm. He was alert and reported no issues with work and daily activities, scoring 28/30 on the MMSE with two points lost for short-term memory. Cranial nerve examination was normal apart from subtle rotatory nystagmus looking up and out bilaterally. There was no resting tremor in the upper limbs, but bilateral dystonic inversion of the feet was noted (Supplementary Video 1 in the online-only Data Supplement). Repetitive finger taps and finger movements were mildly bradykinetic bilaterally. Repetitive heel and foot taps were moderately slowed bilaterally, and this feature was more evident on the left. Tone and power were both preserved throughout the upper and lower limbs. There were no appendicular cerebellar signs. Sensory examination revealed bilateral reduced vibration to the level of the ankles distally and impairment of joint proprioception in the great toes. Reflexes were absent with reinforcement in both the upper and lower limbs, and plantar responses were flexor. In the upright position, occasional high amplitude rhythmic tremor was noted in the right leg. His gait was broad-based. After 20 meters, the dystonic foot inversion was accentuated, and the right toe became extensor. His tremor abated in the supine position. He was unable to perform tandem walking but could walk on heels and toes with hand-holding support for balance. Romberg’s test was positive.
Nerve conduction studies and somatosensory evoked potentials revealed evidence of moderate to severe generalized sensory length-dependent axonal polyneuropathy. Investigations for metabolic, infectious, and immune-mediated neuropathies and movement disorders were unremarkable with bland cerebrospinal fluid and no abnormalities on MRI brain and whole spine. Muscle biopsy and assessment for mitochondrial depletion or multiple deletions were not performed.
A trial of low-dose levodopa/benserazide uptitrated to 300 mg of daily levodopa was performed with a significant improvement in tremor and dystonia (Supplementary Video 1 in the online-only Data Supplement) without significant side effects, and the patient was able to continue full-time work. However, in the last four years, he developed incipient progressive external ophthalmoplegia; a resting tremor in the right upper limb; occasional myoclonic jerks in the shoulder, arm, forearm and distal fingers; appendicular cerebellar dysfunction; and mild bilateral foot drop.
The patient underwent whole-genome sequencing as reported [6]. Although not detected on initial analysis, two POLG variants were later established as disease-relevant following clinical and genetic re-evaluation: NM_001126131.2:c.2209G>C (p. Gly737Arg) (pathogenic, PS1, PM2, PP2, PP3, PP5) as previously reported [7,8] and a variant not previously associated with human disease, NM_001126131.2:c.3305A>C (p.Gln1102Pro) (likely pathogenic PM1, PM2, PP2, PP3). The POLG variants were shown to affect both alleles in a family study (Figure 1). On review of our whole genome sequencing database from the initial study (n = 111 probands with dystonia), this was the only proband who could be characterized as ‘dystonia and peripheral neuropathy.’
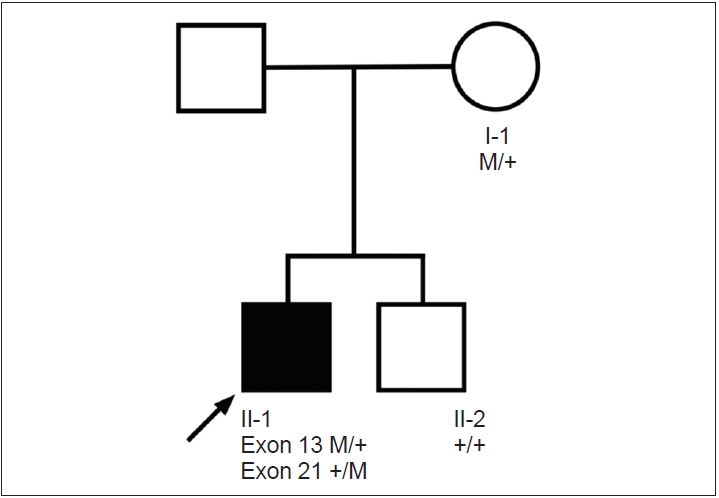
Family pedigree demonstrating biallelic POLG variants. Squares indicate males, circles indicate females, filled symbols indicate affected individuals, and arrow indicates probands. POLG Exon 13, c.2209G>C M/+ Exon 21, c.3305A>C +/M.
DISCUSSION
We report a case of dystonia and peripheral neuropathy caused by compound heterozygous POLG mutations, including a new likely pathogenic variant (p.Gln1102Pro). The known pathogenic variant (p.Gly737Arg) has previously been identified with compound heterozygosity in two siblings with a similar presentation of early-onset parkinsonism, dystonic toe curling, action tremor, bradykinesia and a sensorimotor axonal peripheral neuropathy in their twenties with partial response to dopamine [8] in conjunction with another known pathogenic variant of POLG.
This is one of only a few instances whereby dystonia was reported as a prominent manifestation of a POLG-related disorder. It is now apparent that POLG mutations can manifest with a wide range of movement disorders (Table 1). Differentials for “dystonia with peripheral neuropathy” include Refsum disease and PLA2G6-associated neurodegeneration. However, peripheral neuropathy is a well-established manifestation of POLG mutations and may initially present as Charcot-Marie-Tooth Disease (CMT). In fact, a recent study reported a patient presenting with CMT with one of the same mutations described in this case (p.Gly737Arg) [7].
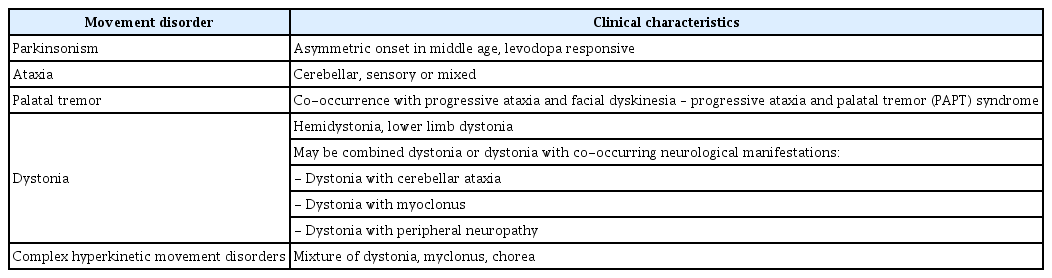
Movement disorders associated with POLG mutations
This report provides further evidence that the phenotype of “dystonia with peripheral neuropathy” may be due to a POLGrelated disorder [9]. Notably, from our original whole genome sequencing study, this proband was the only one identified to have both peripheral neuropathy and dystonia out of 111 probands with heterogeneous dystonia phenotypes. Other dystonia phenotypes of POLG mutations include “dystonia with cerebellar ataxia” and “dystonia with myoclonus,” [9] so POLG-related disorders should also be considered more broadly as a cause of dystonia with cooccurring neurological manifestations.
In this case, the genetic diagnosis was made prior to the appearance of readily recognizable features of mitochondrial disease such as progressive external ophthalmoplegia. Of importance, the diagnosis had implications for further investigations and management of this patient. The identification of POLG mutations as the genetic cause obviated the need for invasive investigations, such as muscle or nerve biopsies, which may have posed additional harm to the patient. The diagnosis may also guide the prescription of medications in the future, as we can avoid medications that are known to exacerbate POLG-related disorders, such as sodium valproate, which can precipitate liver failure [1]. Sodium valproate may be considered in the eventuality of a seizure disorder, a known complication of POLG mutations. Furthermore, our case showed a clear response to levodopa therapy. A sustained long-term response to levodopa has also been previously described in a case of dystonia with autosomal dominant progressive external ophthalmoplegia type 1 [5], emphasizing the importance of the use of levodopa in patients with dystonia-related POLG mutations.
Finally, we highlight the benefit of periodic re-evaluation of genomic data, which can reveal additional diagnoses over time, providing long-term future value for patient healthcare [10].
In summary, we highlight POLG mutations as an exemplar of the “dystonia with peripheral neuropathy” phenotype and that the dystonia in POLG-related disorders may be levodopa responsive. Clinicians should consider POLG mutations when presented with this combination of neurological manifestations, particularly given that this may have implications for the further investigations and management of their patients.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.20159.
Supplementary Video Legends
Video 1. Pre-levodopa assessment in a patient at age 30 revealed dystonic inversion of the feet, a striatal toe accentuated by exertion and impaired balance. The patient had an asymmetric tremor isolated to the lower limbs, and bradykinesia was present bilaterally on repetitive finger and foot movements. Joint proprioception was also impaired distally. An improvement was noted post-levodopa.
Notes
Ethics Statement
The study received ethics approval from the Human Research Ethics Committee affiliated with the Northern Sydney Local Health District, New South Wales Government, Australia (HREC reference number RESP15/314) and the ethics committee affiliated with the Royal Prince Alfred Hospital, Sydney Australia (HREC reference number 13/RPAH/363). Written and informed consent was obtained for the written case report and video to be published.
Conflicts of Interest
The authors have no financial conflicts of interest.
Author Contributions
Conceptualization: Michael W. Hayes. Data curation: Kishore Raj Kumar, Michael W. Hayes, Jessica Qiu, Eloise Watson. Formal analysis: Kishore Raj Kumar. Investigation: Kishore Raj Kumar. Methodology: Kishore Raj Kumar. Project administration: Michael W. Hayes, Kishore Raj Kumar, Jessica Qiu. Supervision: Michael W. Hayes, Kishore Raj Kumar. Writing—original draft: Jessica Qiu, Kishore Raj Kumar. Writing—review & editing: all authors.
Acknowledgements
Kishore Raj Kumar receives funding to study dystonia from the Paul Ainsworth Family Foundation, and receives a Working Group Co-Lead Award from the Michael J. Fox Foundation, Aligning Science Across Parkinson’s (ASAP) initiative. We thank the patient presented in this report for his written permission to publish this case and the associated video.