Manganese and Movement Disorders: A Review
Article information

Abstract
Scientific and technological advances achieved with industrial expansion have led to an ever-increasing demand for heavy metals. This demand has, in turn, led to increased contamination of soil, water and air with these metals. Chronic exposure to metals may be detrimental not only to occupational workers but also to the nonoccupational population exposed to these metals. Manganese (Mn), a commonly used heavy metal, is an essential cofactor for many enzymatic processes that drive biological functions. However, it is also a potential source of neurotoxicity, particularly in the field of movement disorders. The typical manifestation of Mn overexposure is parkinsonism, which may be difficult to differentiate from the more common idiopathic Parkinson’s disease. In addition to environmental exposure to Mn, other potential etiologies causing hypermanganesemia include systemic health conditions, total parenteral nutrition and genetic mutations causing Mn dyshomeostasis. In this review, we critically analyze Mn and discuss its sources of exposure, pathophysiology and clinical manifestations. We have highlighted the global public health impact of Mn and emphasize that movement disorder specialists should record a detailed social and occupational history to ensure that a toxic etiology is not misdiagnosed as a neurodegenerative disease. In the absence of a definite therapeutic option, early diagnosis and timely institution of preventive measures are the keys to managing its toxic effects.
Metals are found in the Earth’s crust not only from natural sources such as volcanic eruptions and weathering of rocks but also from anthropogenic activities such as industrialization, urbanization and mining [1]. These metals have contaminated the air, soil and water with toxic concentrations that are increased by several fold, jeopardizing human health. Heavy metals have a specific density greater than 5 g/cm3 and include essential and nonessential metals [2]. Essential metals serve as cofactors for the enzymes required for protein synthesis, redox reactions, electron transport and protein and carbohydrate metabolism [3,4]. These essential elements are required in the human body in trace quantities ranging from 50 μg to 18 mg per day to maintain normal homeostasis [5]. Overexposure to heavy metals leads to dysfunctional homeostasis when they compartmentalize in preferential cells and bind to proteins and nucleic acids, leading to a disruption of cellular functions. In the field of movement disorders, manganese (Mn) is notorious for causing parkinsonism and its toxicity must be differentiated from the more common idiopathic Parkinson’s disease (IPD). Unlike IPD, which targets the substantia nigra pars compacta (SNpc), Mn preferentially targets the globus pallidus and affects SNpc to a lesser extent [6]. Although the phenomenology of the movement disorders between the two diseases differs, a high degree of clinical acumen is required, along with a detailed history of the duration and type of toxic exposure causing Mn-induced parkinsonism. From a clinical perspective, this history is important, as the management of Mn-induced parkinsonism is different from neurodegenerative dystonias and parkinsonism. Altered Mn metabolism may also occur due to impaired excretion in patients with systemic diseases and mutations in the genes responsible for the uptake of Mn across the cell membrane. These disorders also result in movement disorders secondary to elevated Mn levels in the blood and brain. The approach to the management of these groups of disorders differs from disorders caused by environmental and occupational exposure.
In this review, we will discuss the biology of Mn, its public health impact, pathophysiological basis and management of the movement disorders related to Mn toxicity.
Manganese
Manganese (Mn) serves as a cofactor for the enzymes glutamine synthetase, pyruvate carboxylase and superoxide dismutase (SOD), which are necessary for energy metabolism and antioxidant function [7]. Mn is naturally present as a component of other minerals, and the most common Mn-bearing mineral is pyrolusite (MnO2) [8]. Forest fires and volcanic eruptions are the major atmospheric natural sources of Mn. Major industrial sources of Mn include Mn ore mining, alloy production, welding, dry alkaline battery manufacturing, ceramic use, fungicides and as an antiknock agent (added to gasoline to increase the temperature and pressure for autoignition) [9]. As an essential metal, Mn is required only in trace amounts, with an oral recommended intake of approximately 2.0 mg/kg, with the highest levels present in legumes, nuts, rice and whole grains. Only 3–5% of ingested Mn is absorbed through the gastrointestinal tract and is sufficient for essential functions [10].
Manganese uptake and metabolism
The mechanisms of Mn uptake are not clear, although the divalent metal transporter 1 (DMT1) probably aids in Mn absorption by enterocytes (Figure 1). The metal importer SLC39A14 (Solute Carrier Family 39 member 14) and exporter SLC30A10 (Solute Carrier Family 30 member 10) transport Mn across the intestine. Mn levels are maintained by the strict regulation of intestinal absorption, metabolism in the liver and excretion via bile [6,11]. SLC30A10 is expressed in the liver as well and exports Mn into bile [12]. Mn circulates in the body mainly in the more stable divalent Mn2+ and trivalent Mn3+ states. The pathways of Mn entry into the brain include the capillary endothelial cells of the blood-brain barrier, the cerebrospinal fluid and the olfactory nerve through the nasal cavity [13]. Mn2+ readily reaches the brain either as a free ion or protein-bound form, while Mn3+ enters the nervous system by transferrin receptor (TfR)-mediated mechanisms [6]. Divalent intracellular Mn is transported into the mitochondria via the calcium uniporter and is the primary pool of the metal used for essential purposes [9]. Elimination of excess Mn occurs through conjugation with bile by the liver followed by fecal excretion. Chronic diseases of the liver impair the uptake and excretion of Mn and are associated with Mn toxicity, as discussed later. Limited amounts of Mn are also excreted in sweat, milk and urine [14]. The latter route becomes important in patients with renal diseases, where Mn levels are either elevated or decreased due to multiple factors, such as associated iron-deficiency anemia, protein energy malnutrition and dietary restrictions [15,16]. Patients with chronic renal failure in the predialysis phase were found to have high serum Mn levels, while studies of patients undergoing hemodialysis have reported low Mn levels [15,16]. However, brain abnormalities on magnetic resonance imaging and parkinsonism have been reported in patients receiving hemodialysis [17].
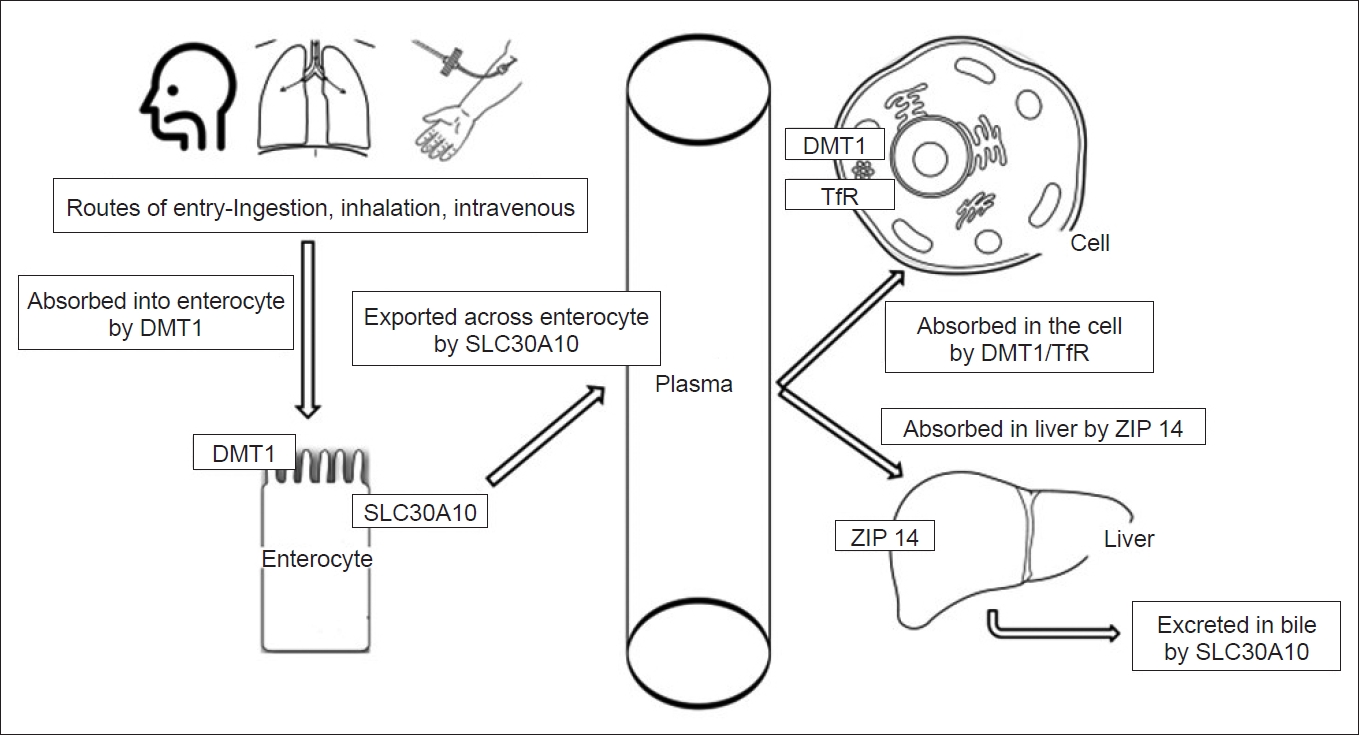
Uptake and transport of Mn. ZIP 14/SLC30A10: metal transporters. DMT1: divalent metal transporter 1, Mn: manganese, TfR: Transferrin receptor.
Manganese toxicity and public health impact
John Couper first reported Mn-induced neurotoxicity in 1837 in a bleach factory, although the harmful systemic effects of the metal were known even earlier [18]. The toxic effects of Mn are related to its organic and inorganic compounds. Organic compounds such as methylcyclopentadienyl Mn tricarbonyl (MMT) have been used since the 1970s as octane improvers of gasoline at concentrations of 8.3 mg of Mn/L of gasoline. The combustion of MMT releases inorganic phosphate and sulfate salts that are primarily responsible for the toxic effects. These inorganic salts, such as Mn chloride (MnCl2), Mn sulfate (MnSO4) and Mn phosphate (MnPO4), contain Mn in the Mn(II), Mn(III), or Mn(IV) oxidation states and are directly toxic to critical targets [19]. The European Commission methodology issued a statement in 2013 regarding the risk assessment of MMT as a fuel additive in petrol and stated that an MMT treatment rate in the range of 8.3 mg Mn/L to 18 mg Mn/L delivers economic benefits without detrimental health effects [20].
Chronic environmental or occupational exposure is primarily observed in miners, welders, smelters, workers in dry-cell battery production, ferroalloy steel plants, glass and ceramics manufacturing industries, and match and fireworks industries [21]. In 2017, the global production of Mn ore, which is essential for the production of iron and steel, was 20 million metric tons. South Africa, followed by Australia, China, Gabon and Brazil, are the largest Mn producers in the world [22]. Along with mining and industrial discharges, Mn is also released into drinking water naturally due to the weathering of rocks and forest fires [23]. Welding fumes exert the most detrimental effects, and with five hundred thousand full-time welders in the United States of America and five and a half million in Europe, substantial numbers of workers are exposed to potential toxic effects of Mn [24]. Numerous studies conducted across the globe have addressed the neurological side effects of Mn exposure on subjects working in the mining industry and smelting or welding occupations. Neurocognitive deficits were noted in patients exposed to Mn in Sweden, Belgium, Singapore, South Africa and Italy, as well as other countries. All these studies reported poor performance on neurocognitive tests and neurological dysfunction (verbal fluency, increased reaction time, reduced motor speed, visuomotor processing, eye-hand coordination and hand steadiness) in participants exposed to chronic Mn intoxication [25-29]. Roels et al. [26] calculated the lowest ‘permissible dose’ for individuals with Mn exposure to prevent neurological deficits of approximately 750 pg/m3 per year for respirable dust. In a recent study of a welder cohort from Pennsylvania, even low levels of chronic Mn exposure produced low diffusion tensor fractional anisotropy in the basal ganglia associated with movement disorders [30]. Mn levels in the soil and groundwater may increase, particularly in the vicinity of ferroalloy plants and industries, and expose neighboring communities to detrimental health effects. The toxic effects of drinking water contaminated with higher than the recommended concentration of Mn (not more than 400 μg/L as per WHO guidelines) have been observed worldwide, affecting countries such as Bangladesh, Italy and Canada [11]. The use of MMT in Canada, the USA, France, Argentina and Australia with high traffic density and chronic exposure to the dithiocarbamate fungicides maneb and mancozeb are other sources of Mn toxicity [31,32]. A community in Quebec near a former Mn alloy production plant was studied to determine the health effects of Mn. Older individuals presented an inverse relation between blood Mn levels and psychomotor slowing and a direct effect on mood symptoms and performance on tests of auditory recall and visual recognition. This study was the first to evaluate the negative health effects of Mn on individuals with nonoccupational exposure to high Mn levels in the environment [33]. Similarly, studies from communities in the vicinity of Mn ferroalloy plants suggested that prolonged environmental exposure to Mn may increase the risk of parkinsonism and postural disturbances [34,35]. At comparable exposures, children are at increased risk of Mn toxicity because they have better absorption and a lower body mass [36]. Developmental delay and low intelligence quotient scores have been documented in children exposed to high Mn concentrations. In a pooled analysis of children exposed to Mn in drinking water from the provinces of Quebec and New Brunswick in Canada, Kullar et al. [37] reported an association between a lower IQ score and higher Mn levels.
Other susceptible populations at risk
Total parenteral nutrition solutions contain 7.3 μg/L Mn without additional supplementation. Additionally, further Mn supplementation and the fact that intestinal absorption is bypassed may lead to toxic accumulation in the brain [38]. Abdalian et al. [39] evaluated Mn levels in 16 patients on TPN with mean daily Mn supplementation within the American Medical Association Nutritional Advisory Group Guidelines (150–800 µg/d) and found that the high blood Mn levels in these patients correlated with changes in the pallidum signal on magnetic resonance brain imaging. This finding is a cause for major concern, as most patients on TPN are in long-term care homes without proper monitoring and present neuropsychiatric issues. Updated guidelines from the American Society for Parenteral and Enteral Nutrition recommend lowering the recommended Mn levels to 60–100 µg/d.
In Turkey, intravenous use of a ‘Russian cocktail’ (psychostimulant containing ephedrine, acetylsalicylic acid and potassium permanganate) was associated with Mn toxicity-induced parkinsonism in seven patients. Mn toxicity resulted from the large amounts of potassium permanganate used to disinfect the solution [40].
Approximately 1–2% of patients with chronic liver disease have acquired hepatocerebral degeneration (AHD) [41]. Two- to seven-fold increases in Mn deposition in the pallidum have been observed in patients with AHD secondary to impaired biliary excretion or portosystemic shunts [42].
Autosomal recessive mutations in the Mn exporters SLC30A10 and SLC39A14 increase the susceptibility to Mn toxicity and cause adult onset dystonia parkinsonism and childhood developmental disorders, respectively [43,44]. Finally, iron deficiency anemia predisposes individuals to an increased Mn burden, as the common transporters for the uptake of these metals (DMT1) are upregulated in these individuals, promoting increased Mn uptake [45].
The causes of hypermanganesemia are shown in Figure 2.
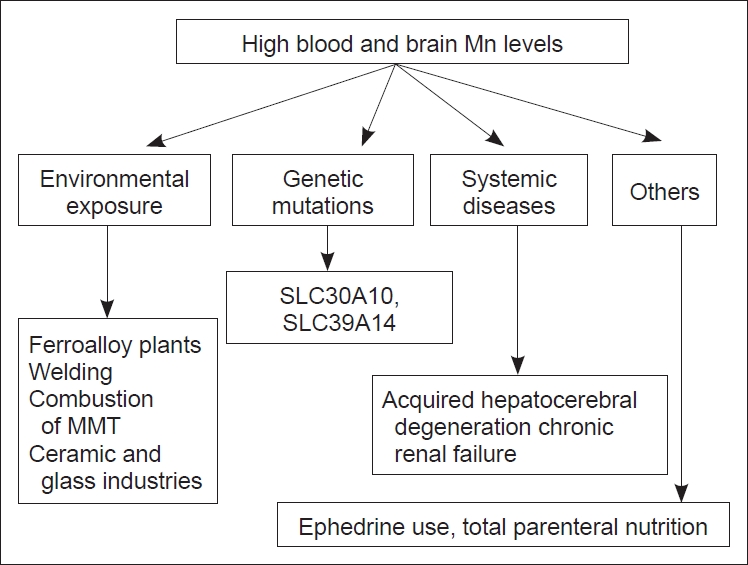
Different modes of hyper manganesimia. MMT: methylcyclopentadienyl Mn tricarbonyl, Mn: manganese.
Movement disorders and manganese
Neurotoxicity due to Mn exposure causes neuronal loss and gliosis in the basal ganglia, with the globus pallidus, the substantia nigra pars reticulata, and the striatum identified as particularly vulnerable structures [46]. DMT1 is expressed at high levels in the striatum, pallidum and substantia nigra, consistent with the sites of Mn accumulation [6]. The specific involvement of dopamine-rich regions and the pallidum has been attributed to the dopamine transporter (DAT) that binds Mn and deposits it in dopamine-rich regions. In DAT knockout mouse models, significantly less accumulation of Mn in the striatum was observed compared to wild-type mice [47]. The authors speculated that increased Mn deposition in the pallidum compared to the striatum might be due to axonal transport of Mn from the striatum to the pallidum [48]. In addition to the basal ganglia, other regions affected by Mn toxicity include the cerebellum, red nucleus, pons and anterior horn cells of the spinal cord [11,49].
Molecular mechanisms of Mn toxicity
Mitochondrial dysfunction, along with protein misfolding and neuroinflammation, are hypothesized to reflect Mn toxicity (“neurotoxic triad”) [50]. Mn is sequestered in the mitochondrial matrix, where it inhibits calcium efflux, leading to calcium accumulation. Elevated matrix calcium levels increase the formation of reactive oxygen species, mediating the toxic effects of Mn [51,52]. An autopsy study of South African Mn mine workers showed a higher microglial density in the globus pallidus and a lower astrocyte:microglia ratio in the caudate, putamen and globus pallidus. Neurodegeneration related to Mn may initially lead to microglial infiltration in the basal ganglia and astrocyte damage with subsequent neuronal loss [53]. Mn has been proposed to facilitate the aggregation of α-synuclein and its prionlike intercellular transmission, which leads to the development and progression of parkinsonism in individuals with chronic exposure [54]. Mn is a cofactor for the enzyme glutamine synthetase (GS), which is expressed mainly in astrocytes and converts glutamate to glutamine. Reduced activity of GS in individuals with chronic Mn exposure thus promotes increased glutamatergic signaling. In addition, chronic Mn intoxication decreases the levels of glutamate:aspartate transporter, which is primarily responsible for synaptic glutamate clearance via astrocytes. These two mechanisms have been proposed to explain glutamatergic excitotoxicity and subsequent neuronal loss in patients with manganism [22].
Clinical features of manganese toxicity
Manganism
Chronic exposure to Mn leads to ‘manganism’, the complex syndrome of Mn neurotoxicity characterized by cognitive, behavioral and extrapyramidal dysfunction. Psychiatric symptoms predominate in the early phase, while an akinetic rigid syndrome with minimal tremors, axial and appendicular dystonia and ‘cock walk’ (discussed later) appears in the established phase [55]. Although Mn exposure causes extrapyramidal dysfunction, the clinical features of Mn toxicity are rather different from those observed in patients with idiopathic Parkinson’s disease. While PD is a disease of advancing age, Mn-related parkinsonism affects the younger population exposed to Mn in various occupations [56]. Manganism starts gradually with psychiatric symptoms (Mn madness or locuramanganica) followed by motor abnormalities, while behavioral and psychiatric abnormalities appear later in a typical patient with PD [57]. The symmetrical onset of symptoms, action rather than resting tremor and a poor response to dopaminergic therapy are helpful for distinguishing manganism from PD [57,58]. Eight patients described by Josephs et al. [58] had varying presentations, including parkinsonism, multifocal myoclonus, cognitive dysfunction, sleep disorders and vestibular dysfunction. This clinical heterogeneity in the presentation further reiterates the need for recording a detailed social and occupational history to evaluate the possible sources of Mn exposure in patients presenting with features atypical for idiopathic PD. The ‘cock walk’ described earlier is the characteristic finding in which patients strutting on the toes without the heels touching the floor, with an erect spine and flexion at the elbows due to the generalized dystonic posture. This fast stepping gait mimics how chicken run. This gait contrasts with the typical shuffling gait observed in patients with idiopathic PD. However, this characteristic gait was not observed in a study by Andruska et al. [57] evaluating over 4,000 Mn-exposed workers. The authors postulated that the duration and quantum of exposure results in a wide range of symptoms, ranging from mild tremor and bradykinesia to severe hypokinesia and the peculiar gait dysfunction [57]. Interestingly, ten years after the cessation of exposure, patients continued to experience a progression of their parkinsonian symptoms despite the absence of signal changes on brain imaging. Thus, Mn causes long-term progression that affects the globus pallidus and substantia nigra pars reticulata, although the mechanism of this degeneration years after exposure remains unknown [59]. Notably, this cock-walk gait phenotype has also been reported in individuals with spinocerebellar ataxia type 3 (SCA3) and pantothenate kinase-associated neurodegeneration (PKAN) due to associated dystonia [60,61]. In comparison to individuals with IPD, patients with parkinsonism from manganism are more likely to have 1) less frequent and atypical tremor, 2) more frequent dystonia (e.g., facial grimacing and cock walk), 3) a greater tendency to fall backwards early, 4) poor or ill-sustained response to levodopa, and 5) an absence of reduced striatal fluorodopa uptake in a DAT scan.
Although parkinsonism is the most common clinical presentation of Mn toxicity, Ghosh et al. [62] recently reported Mn toxicity in a tea seller who presented with cognitive dysfunction and choreiform movements following the consumption of tea with a high Mn concentration. Likewise, another patient with chronic Mn overexposure presented myoclonic jerks without parkinsonism [63]. These isolated case reports expand the spectrum of movement disorders observed in individuals with Mn toxicity and emphasize the importance of recording a detailed social and occupational history.
Acquired hepatocerebral degeneration
In patients with acquired hepatocerebral degeneration (AHD), toxic substances, primarily Mn, accumulate due to dysfunctional clearance. Portosystemic shunts are a predisposing factor for neurological toxicity, and these patients may have normal liver enzyme levels [41]. Neurological symptoms are unrelated to the duration of the hepatic pathology and consist of parkinsonism, ataxia or myelopathy. Parkinsonism, unlike IPD, is symmetric, with gait abnormalities, a postural tremor, variable levodopa response and characteristic absence or minimal resting tremor [41]. However, resting tremor has been reported as a common finding in other studies [64,65]. Other movement disorders, such as chorea, dystonia and hemiballismus, have also been reported in individuals with AHD. Ataxia is observed in 30% of patients and usually occurs in association with extrapyramidal features. Mild cognitive abnormalities with inattention and psychomotor slowness are common features [41,64,65]. The disorder is differentiated from Wilson’s disease by the absence of Kayser-Fleischer rings, a variable age at onset and normal laboratory findings of serum ceruloplasmin and 24-hour urinary copper levels. Patients who are exposed to Mn from exogenous sources usually have more compulsive behaviors with normal liver functions and normal serum ammonia levels than patients with AHD.
Gene mutations and Mn-related clinical disorders
In 2012, Tuschl et al. [66] reported an autosomal recessive mutation in the Mn transporter SLC30A10 that caused Mn accumulation and a syndrome characterized by early onset dystonia, cirrhosis and polycythemia. Subsequently, more families with the same mutations were identified, but the clinical presentation is quite variable and includes spastic paraparesis without dystonia, late onset parkinsonism with postural instability and isolated hepatomegaly. An evaluation of blood samples from these patients shows high serum Mn levels with polycythemia and low serum ferritin levels [43,67].
SLC39A8 (Solute Carrier Family 39 member 8) is a member of the solute carrier gene superfamily that transports Mn, zinc, cadmium and iron across the plasma membrane. p.GLY38Arg gene mutations in SLC39A8 impair the transport of these metals across the plasma membrane, resulting in extremely low blood zinc and Mn levels. The mutation has been identified in 2 children of Egyptian descent and 6 Hutterite children (ethno-religious community in Canada). These children presented a syndrome characterized by developmental delay, hypotonia, strabismus and cerebellar atrophy on MRIs of the brain [44].
SLC39A14, a member of the same solute carrier family, facilitates Mn uptake in the liver for excretion into bile. Mutations in this gene lead to impaired hepatic uptake of Mn and excess Mn in the brain, leading to rapidly progressive dystonia with variable parkinsonian signs. Unlike SLC30A10 mutations, signs of liver failure and polycythemia are not observed in these individuals [68].
A comparison of the clinical presentations of patients with these three gene mutations causing altered Mn levels is summarized in Table 1. Prominent causes of high brain Mn levels along with the clinical manifestations are outlined in Table 2.

Gene mutations linked to Mn metabolic pathways

Clinical manifestations of Mn toxicity linked to various causes
Imaging in patients with manganese-related movement disorders
Due to the paramagnetic properties of Mn, its deposition in the pallidum is detected as hyperintensities in the globus pallidus on magnetic resonance imaging (MRI) and is a characteristic MRI finding in the brain [39]. The pallidal index (PI) is the ratio of the signal of the globus pallidus to the frontal white matter, and an enhanced PI is a reliable marker of Mn exposure, even in the asymptomatic stage of the disease [11]. The hyperintensities are symmetric and may extend to the adjacent putamen and mesencephalon, particularly in individuals with AHD [64]. In individuals with AHD, apart from this classical finding, patients with an ataxia presentation have atypical findings of symmetrical hyperintensities in the cerebellum with variable degrees of cortical atrophy that are not detected in individuals with manganism [64,65]. Dopaminergic neuronal function is typically assessed by performing a dopamine transporter scan (DAT scan), which shows reduced and asymmetric binding in the striatum of patients with idiopathic PD, whereas in individuals with Mn-induced parkinsonism, the DAT scan is mostly normal or reveals a slight decrease in dopamine uptake [56]. The abnormality in the DAT scan of patients with Mn-induced parkinsonism may not be related to degeneration of the nigrostriatal dopaminergic terminal (as in individuals with IPD) but to an interaction of Mn with striatal radioligand binding to DAT [69]. Positron emission tomography (PET) scanning is another noninvasive functional imaging modality that reflects neuronal aromatic L-amino acid decarboxylase activity and the state of dopaminergic presynaptic nerve terminal function. This imaging modality has reported sensitivities and specificities of 90–100% and 91%, respectively, in IPD diagnosis [70]. In a patient with end-stage liver disease, a 3,4-dihydroxy-6-[F] fluoro-L-phenylalanine (18F-DOPA) PET scan showed reduced uptake in the caudate compared to ten patients with IPD, where uptake in the posterior putamen was significantly reduced [71]. Criswell et al. [72] compared the FDOPAPET findings from welders, Mn-exposed workers (working around welding fumes with less than 500 hours of lifetime exposure) and nonexposed controls. They found reduced uptake in the caudate nucleus in the welders and Mn-exposed workers compared to the controls, along with a reduced caudate/putamen uptake ratio. Low uptake was observed even in individuals with a normal Unified Parkinson’s Disease Rating Scale (UPDRS) part 3 motor score. Their results are in sharp contrast to the reduced uptake predominantly observed in the posterior putamen of patients with IPD. Additionally, preferential effects of Mn toxicity on the caudate might explain the cognitive deficits that predominate the clinical characteristics of patients with Mn-induced movement disorders.
Treatment of manganese-related movement disorders
In contrast to idiopathic PD, levodopa does not exert a beneficial effect on manganism [66]. This finding prompted clinicians to attempt chelation to remove the excess metal deposited in the brain and determine whether the clinical scores of patients with Mn toxicity would improve. Upon chelation with intravenous ethylenediaminetetraacetic acid (EDTA), free Mn excretion is increased in the urine with a reduction in blood Mn levels [9]. Herrero Hernandez et al. [73] treated seven welders affected by manganism with 2 g of calcium disodium EDTA (CaNa2EDTA). Four patients experienced a reduction in their symptoms, while tremors were partially reduced in one patient. The improvement in clinical symptoms occurred concomitant with a radiological improvement. The authors concluded that the early institution of therapy prior to the establishment of neural damage is the key to successful chelation in individuals with manganism. However, EDTA is highly water soluble with poor penetration across the blood brain barrier; hence, its role in ameliorating the neurotoxic effects of Mn is questionable. Para-amino salicylic acid (PAS) and its metabolite, N-acetyl paraamino salicylic acid (Ac-PAS), both bind Mn and remove it from its excess stores. Both PAS and Ac-PAS have been detected in the brain and hence may be a potential chelation option for patients with manganism [11]. In an experimental study, Yoon et al. [74] observed that PAS blocks Mn-induced apoptosis in the human neuroblastoma cell line SK-N-MC. Jiang et al. [75], administered fifteen courses of 6 gm of PAS over a course of 4 months to a fifty-year-old patient with parkinsonism secondary to chronic Mn exposure. At the 7-month follow-up, her symptoms persisted, but at the 17-year follow-up visit, she had a mild hand tremor and gait abnormality, but other symptoms of generalized tremors, tetany, and severe gait abnormalities had disappeared. Researchers have postulated that PAS can remove both Mn2+ and Mn3+ ions; however, the exact chemical interaction between PAS-Mn is largely unknown. Although animal studies have shown a reduction in Mn levels after the initiation of chelation therapy, a paucity of literature supports chelation as a treatment option for manganism. In the absence of effective therapeutic options and a poor response to levodopa, chelation should be attempted in patients with manganism, particularly those diagnosed early and presenting with mild symptoms.
In contrast, favorable outcomes have been reported in patients with AHD after liver transplantation. Compared to medical management, three living donor liver transplant recipients experienced a resolution of their clinical and radiological symptoms at the sixteen-month follow-up [76]. Similar reports of persistent long-term clinical benefits suggest that liver transplantation is the intervention of choice in these groups of patients [77,78]. However, not all centers have this type of facility, and the associated comorbidities may make patients unsuitable candidates for surgery. In these scenarios, some success has been obtained with rifaximin and attempted chelation with trientine [41,79].
Stamelou et al. [67] showed a reduction in the globus pallidus signal intensity with an improvement in dystonia in a patient with a SLC30A10 mutation presenting with hypermagnesemia who was treated with CaNa2EDTA and followed for more than 10 years. 2,3-Dimercaptosuccinic acid or D-penicillamine are other alternatives, and iron supplementation alone has reduced Mn levels in these patients [68]. Chelation with CaNa2EDTA has achieved variable success in patients carrying SLC39A14 mutations, and the time of onset of chelation and symptom severity are the major determinants of the outcome of medical management [80]. Mutations in SLC39A8 are associated with Mn deficiency, and oral Mn supplementation is an effective treatment strategy [68].
Prevention of manganese exposure
The aerosols generated by welding fumes and metallurgical operations are the primary source of Mn exposure, and most preventive policies aim to reduce the inhalation of these toxic fumes. The National Institute of Occupational and Safety Health (NIOSH) recommends a level of 1 mg/m3 of Mn time-weighted average levels [22]. As Mn-associated neurological toxicity is observed after chronic exposure to concentrations well below this level, the American Conference of Governmental Industrial Hygienists (ACGIH) lowered this limit to 0.02 mg/m3 in 2016 [81]. Strict policies have been established to ensure that the levels are maintained within this required upper limit and that occupational exposure to workers is minimized. Dilution ventilation, such as exhaust fans, is not sufficient to prevent inhalation of the toxic fumes generated, and local exhaust ventilation, inbuilt with welding equipment, should be provided to occupational workers. Robotic welding, the use of personal protective equipment including masks and gloves, and regular measurement of air quality should be the norm at the workplace. Workers should be well trained and informed about the health risks and advised to remove and change all clothing before leaving the workplace to prevent household contacts from being exposed to Mn. Workers should be routinely assessed regarding the fitness of protective equipment and their health status with the aim of the timely identification of individuals with early signs of disease to initiate early protective measures. Government bodies, private and public workplace heads and workers must work as a team to create a safe and healthy working environment and obtain the highest productivity while minimizing the health hazards [82,83]. While tap and bottled water are generally safe and contain low Mn levels, people living close to industrial sites should be cautious when using well water for consumption, and the water should be regularly analyzed to ensure that Mn levels are below those established by protective agencies. As stated above, iron deficiency promotes increased Mn absorption; hence, children should be monitored and managed for iron deficiency and anemia. Areas with high traffic densities, such as those near a busy highway, have higher amounts of Mn in air samples due to the use of MMT in gasoline. Individuals living around these areas should be monitored for any signs of Mn intoxication.
CONCLUSIONS
Mn is widely used in industrial processes, and hence the population directly or indirectly exposed to the toxic effects of Mn is large. Neurologists and movement disorder specialists should consider toxic etiologies for parkinsonism along with degenerative causes. A detailed social and occupational history should be recorded, including the source of access to drinking water, consumption of sea food or living near a potential source of metal toxicity. Patients with manganism and the resulting parkinsonism due to hypermanganesemia respond poorly to levodopa, and the benefits of chelation therapy are limited to findings from animal studies or isolated case reports. In this scenario, prevention, which is better than the cure, signifies the importance of health and safety measures that must be implemented by agencies dealing with Mn at their workplace and the implementation of strict legislative guidelines to reduce environmental contamination secondary to mining and welding activities.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Author Contributions
Conceptualization: Mandar Jog. Supervision: Mandar Jog. Writing—original draft: Dinkar Kulshreshtha, Jacky Ganguly. Writing—review & editing: Dinkar Kulshreshtha, Jacky Ganguly.
Acknowledgements
None.