Successful Pallidal Stimulation in a Patient with KMT2B-Related Dystonia
Article information

Abstract
Although the KMT2B gene was identified as a causative gene for early-onset generalized dystonia, the efficacy of deep brain stimulation (DBS) in KMT2B-related dystonia has not been clearly elucidated. Here, we describe a 28-year-old woman who developed generalized dystonia with developmental delay, microcephaly, short stature, and cognitive decline. She was diagnosed with KMT2B- related dystonia using whole-exome sequencing with a heterozygous frameshift insertion of c.515dupC (p.T172fs) in the KMT2B gene. Oral medications and botulinum toxin injection were not effective. The dystonia markedly improved with bilateral pallidal DBS (the Burke-Fahn-Marsden Dystonia Rating Scale score was reduced from 30 to 5 on the dystonia movement scale and from 11 to 1 on the disability scale), and she could walk independently. From this case, we suggest that bilateral globus pallidus internus DBS can be an effective treatment option for patients with KMT2B-related generalized dystonia.
Recently, with advancement in next-generation sequencing (NGS), new genetic causes of dystonia have been identified, and the KMT2B gene (MIM: 606834) has been identified as a causative gene for early-onset generalized dystonia [1,2]. KMT2B mutation is reported as a relatively common cause of early-onset generalized dystonia, and the prevalence accounts for up to 10% of early-onset generalized dystonia [3]. Recent studies reported improvements after bilateral pallidal deep brain stimulation (DBS) in patients with KMT2B-related dystonia [1,2,4]; however, there is no consensus yet on the efficacy of pallidal stimulation in KMT2B-related dystonia. Herein, we report a case with genetically confirmed KMT2B-related generalized dystonia, with a marked response to DBS of the bilateral globus pallidus internus (GPi).
CASE REPORT
A 28-year-old woman with generalized dystonia for years visited our outpatient clinic. She was born preterm at 9 months, and her development slowly decelerated after birth, resulting in microcephaly, short stature, cognitive decline, and dysarthria. However, she was able to uneventfully manage her daily activities and had a cleaning job at a welfare center. She had no family history of dystonia or other neurological disorders. She developed right laterocollis and right torticollis with a mobile portion at the age of 27, which then slowly spread to her trunk and bilateral arms (Supplementary Video 1 and 2 in the online-only Data Supplement). The dystonic postures rapidly exacerbated, severely affecting her walking and sitting. Although a sensory trick was noted, it was insufficient for recovery to the neutral position. She described some sleep benefits, but there was no definite fluctuation of the dystonia. The Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) score was 30 of 120 on the dystonia movement scale and 11 of 30 on the disability scale. We tried levodopa, baclofen 10 mg tid, trihexyphenidyl 2 mg tid, diazepam 2 mg qid and botulinum toxin injection. However, these methods were not effective in improving dystonic symptoms. Parkinsonism, ataxia, and myoclonus were not combined with dystonia. Although she could walk independently, both the arms, trunk, and neck were in dystonic posture. Regarding cognition, she was formally diagnosed with mild intellectual disability. She scored 58 on the Korean Wechsler Adult Intelligence Scale-IV, particularly scoring in the low percentile range with regard to working memory (0.1 percentile). When she visited our center at the age of 28, her head circumstance was lower than the 1 percentile (48.8 cm).
For the genetic study, NGS was conducted in the patient and her parents for diagnosis. Genomic DNA was extracted from peripheral blood samples of the patient and parents using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Wholeexome sequencing was performed by Macrogen (Seoul, South Korea) and analyzed with the following criteria. First, the sequencing read was aligned to the human reference genome (GRCh37/ hg19) and filtered by the known target genes related to dystonia, with a genotype quality of > 99. Second, variants were narrowed down based on the public population databases, with a minor allele frequency of < 1% and considered ethnicity with Korean population databases, Korean Reference Genome Database (KRGDB, http://coda.nih.go.kr/coda/KRGDB/index.jsp) and in-house databases. With the inheritance pattern of the patient, we finally focused on the strong candidate, which was confirmed by IGV software (https://software.broadinstitute.org/software/igv/), validated by Sanger sequencing, and underwent pathogenicity evaluation according to American College of Medical Genetics and Genomics 2015 guidelines [5].
We identified the autosomal dominant heterozygosity of c.515dupC (p.T172fs), frameshift insertion in exon 3 of the KMT2B gene in chromosome 19 (Figure 1A) in December 2017. This variant has been revealed as a pathologic variant of KMT2B [1]. Her parents did not have mutations related to this gene (Figure 1B). Brain magnetic resonance imaging (MRI) showed no definite abnormal findings associated with KMT2B mutation; bilateral hypointensity of the globus pallidus with a hypointense lateral streak of globus pallidus externa was observed (Figure 2) [1].
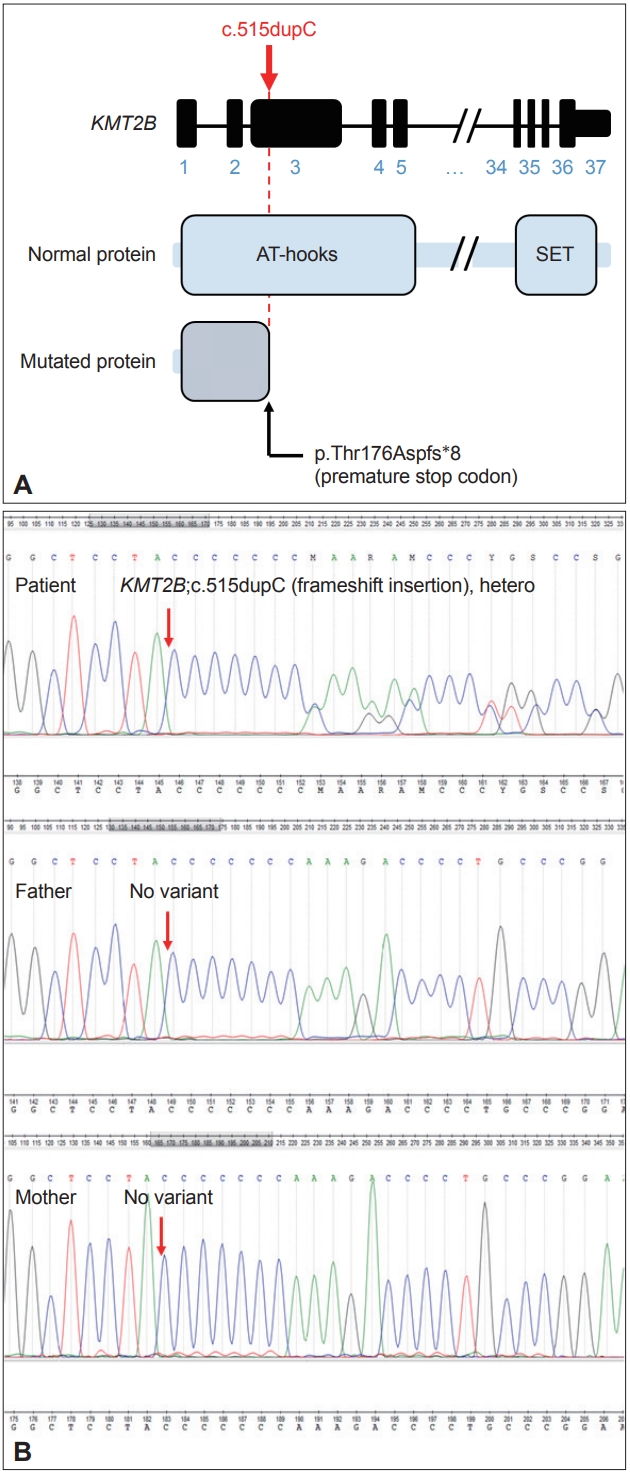
Frameshift insertion mutation in KMT2B gene in the patient with generalized dystonia. A: A schematic diagram of the gene structure of KMT2B indicating the position of frameshift insertion. B: A de novo autosomal dominant mutation in KMT2B. Sequencing chromatograms of the patient and her parents. Sanger sequencing demonstrated a de novo mutation of c.515dupC (p.T172fs), frameshift insertion.
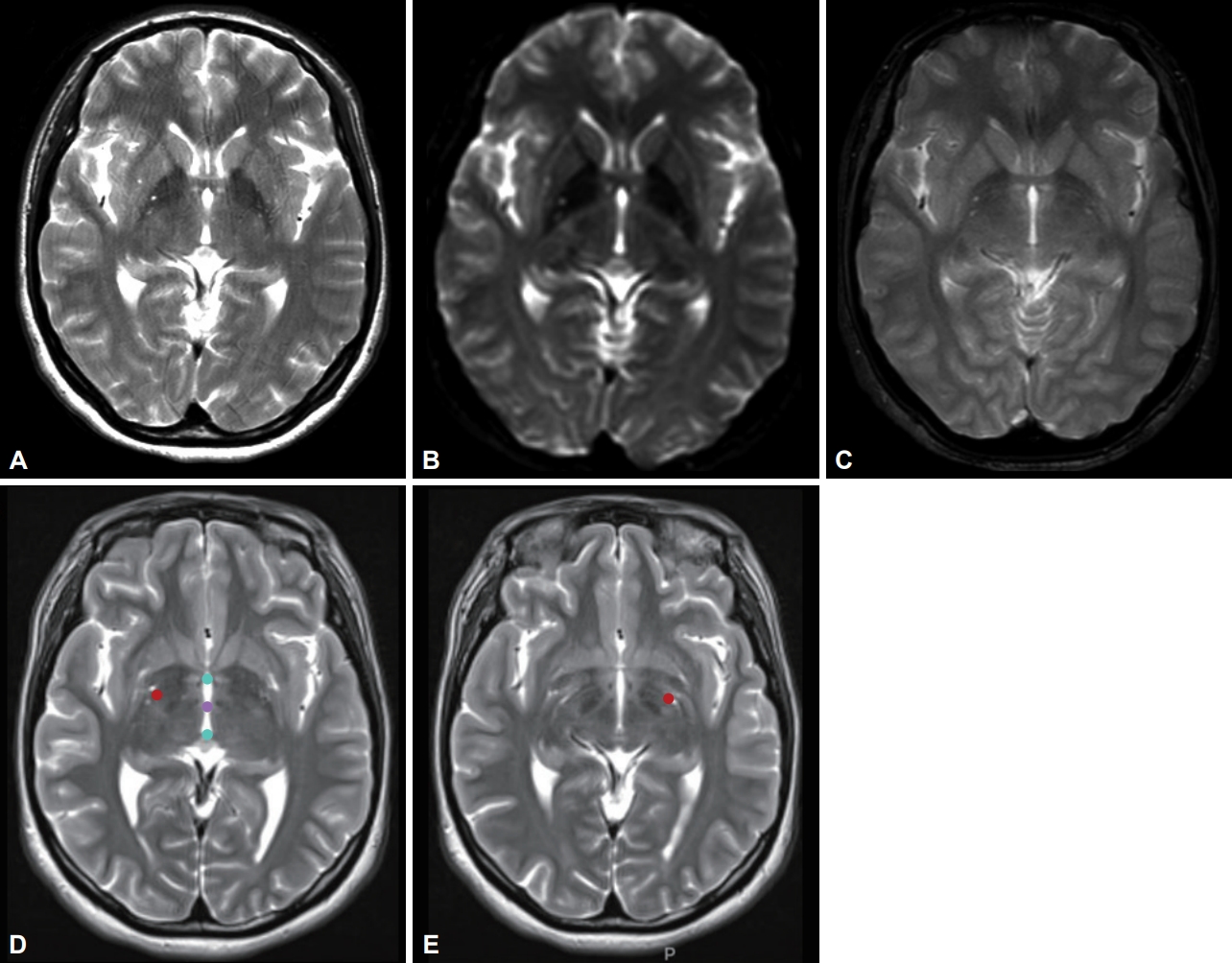
Magnetic resonance imaging (MRI) of the brain shows no abnormal findings specific for the KMT2B mutation. (A) T2-weighted image, (B) echo-planar technique diffusion imaging data with b value of zero, (C) gradient echo sequence. Postoperative fusion MRI images at the level of stimulating contacts on the right side (D) and left side (E) of deep brain stimulation electrodes. Red dots in figures (D) and (E) represent the location of leads. Cyan dots are anterior and posterior commissure points, and purple dot is mid-commissure point.
Bilateral GPi-DBS was performed on this patient. We used directional electrodes because of microcephaly. Abbott InfinityTM 8-channel directional leads (6172, Abbott Neuromodulation, Plano, TX, USA) and an implantable pulse generator (6662, Abbott Neuromodulation) were implanted (Figure 2). One month after surgery, we checked the threshold for efficacy and side effects and chose contacts for stimulation (3A- on the right lead and 2A- on the left). At the outpatient clinic, we slowly increased the stimulation amplitude up to 2.5 mA on the right side and 2.6 mA on the left side, with 140 μs and 160 Hz, for 10 months. Pallidal stimulation slowly improved dystonia in the neck and trunk for 10 months, and her dystonia was stationary until the last follow-up (22 months after DBS). Three months after DBS, she could move her neck and both limbs with almost full range of motion and sit and walk in an almost upright posture. When we evaluated her dystonia 6 months after DBS, the BFMDRS score was dramatically reduced such that the dystonia movement scale was 9 of 120 and the disability scale was 1 of 30 (30 and 11 for movement and disability scale before pallidal DBS, respectively). When we checked the symptoms 22 months after DBS (the last follow-up), the dystonia movement scale was better with 5 of 120.
DISCUSSION
Our patient is the first genetically confirmed dystonia case with a KMT2B mutation in Korea. Additionally, in accordance with previous studies [1,2,4], our patient with the KMT2B mutation showed a significantly good response to pallidal DBS surgery.
KMT2B encodes lysine methyltransferase 2B, which is involved in the methylation of histone H3 at lysine 4 (H3K4). This methylation process is important in the regulation of genetic expression associated with active gene transcription [6]. KMT2B is ubiquitously expressed during brain development and in the adult brain. This expression is highest in the cerebellum and motor control area [1]. According to Meyer et al. [1], KMT2B mutations are predicted to destabilize protein structures. Variants of this mutation substitute the original amino acid with another that alters hydrophobic interactions and surface charge among domains in KMT2B [1,2]. The altered gene and protein expression are thought to be associated with dystonia in KMT2B.
The typical phenotypes were demonstrated in recent studies. The onset age of KMT2B-related dystonia varies according to the subtype of variants. Microdeletion and predicted protein-truncating variants (frameshift, splice site and stop-gain variants) are seen more than missense variants in significantly younger patients (mean age of 4.82 years vs. 11.75 years) [6]. According to a recent cohort study, the onset age of dystonia was 6 years (range, 3–13 years), with a median disease duration of 18 years (range, 3–42 years) [7]. However, atypical cases have also been reported indicating little involvement of the limb, cervical dystonia, and late onset [4]. When compared with previous studies, our patient with heterozygosity of c.515dupC (p.T172fs), frameshift insertion in exon 3 of the KMT2B gene, showed significantly late onset dystonia in her late 20s. Dystonia commonly starts in the lower limbs and involves the cervical, oromandibular, and laryngeal regions [6] and then generalizes over time during the subsequent 4 to 5 years. In our patient, the dystonia started in the cervical region before it became generalized. Compared to previously reported cases, our case showed an older age of onset and shorter time to generalization. Developmental delay, microcephaly, short stature, intellectual disability, psychiatric symptoms, skin lesions, and other systemic symptoms are associated with KMT2B variants. The majority of patients with KMT2B showed the above symptoms in childhood, especially motor symptoms. Our patient also had the above symptoms in her childhood; however, skin lesions and motor symptoms did not develop until her late 20s.
Pallidal DBS is an effective and safe treatment option for dystonia. Before conducting DBS surgery for patients with dystonia, we should consider the clinical features and genotypes. Clinical features for dystonia, such as generalized or segmental involvement and shorter disease duration, are usually regarded as important prognostic factors for pallidal DBS [8]. Apart from the phenotypes, the genotypes are also associated with the outcome after pallidal stimulation in patients with dystonia. Our recent study compared the response to GPi-DBS in patients with DYT1, DYT6, and GNAL mutations [9]. Patients with the DYT1 mutation showed a good response and more reductions in the dystonia motor scale, especially during early follow-up after DBS. However, patients with DYT6 had a lower responder rate and a lower reduction of the dystonia motor scale than those with DYT1. In the GNAL mutation, patients showed good response as DYT1 and sustained better response during late follow-ups than DYT1 and DYT6. However, the exact relationship between the genotypes of dystonia and the outcome from pallidal DBS has not yet been clearly elucidated. Considering the rapid discovery of many genotypes relevant to dystonia using whole-exome or whole-genome sequencing techniques, future studies on these genotypes are warranted.
Additionally, when we chose devices for DBS, we used directional DBS leads with a single-segment monopolar configuration on both GPi (3A- on the right lead and 2A- on the left lead) and a high-frequency stimulation. Considering her microcephaly, more accurate stimulation could be needed and is possible with directional leads within the targeted area.
In conclusion, we report a patient with KMT2B-related dystonia in Korea. Additionally, we also suggest that bilateral GPi-DBS could be an effective treatment option for patients with KMT2B-related dystonia.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.19087.
Supplementary Video Legends
Video 1. The patient shows cervical dystonia on the right side and bilateral arm dystonia and truncal dystonia on the left side.
Supplementary Video Legends
Video 2. After 2 years of bilateral GPi-DBS surgery, the patient shows improvement in the dystonic postures and is able to walk independently.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Author Contributions
Conceptualization: Jong Hyeon Ahn, Minkyeong Kim, Jin Whan Cho, JungIl Lee, Kyung Rae Cho, and Jinyoung Youn. Data curation: all authors. Formal analysis: Jun Kyu Mun, Ah Reum Kim, Jin Whan Cho, Kyung Rae Cho, and Jinyoung Youn. Investigation: Jun Kyu Mun, Ah Reum Kim, Jong Hyeon Ahn, Jin Whan Cho, Kyung Rae Cho, and Jinyoung Youn. Methodology: Jun Kyu Mun, Ah Reum Kim, Jong Hyeon Ahn, Jin Whan Cho, Jung-Il Lee, Kyung Rae Cho, and Jinyoung Youn. Supervision: Jin Whan Cho, Kyung Rae Cho, and Jinyoung Youn. Validation: Jin Whan Cho, Kyung Rae Cho, and Jinyoung Youn. Visualization: Jun Kyu Mun, Ah Reum Kim, and Jong Hyeon Ahn. Writing—original draft: Jun Kyu Mun, Ah Reum Kim, Kyung Rae Cho, and Jinyoung Youn. Writing—review & editing: Jin Whan Cho, Kyung Rae Cho, and Jinyoung Youn.
Acknowledgements
The patient and her parents gave written informed consent.