Progressive Supranuclear Palsy with Predominant Cerebellar Ataxia
Article information

Abstract
Progressive supranuclear palsy (PSP) is characterized by supranuclear gaze palsy, dystonic rigidity of the neck and upper trunk, frequent falls and mild cognitive impairment. Cerebellar ataxia is one of the exclusion criteria given by the National Institute of Neurological Disorders and Stroke and the Society for Progressive Supranuclear Palsy. As a result, pathologically proven PSP patients exhibiting cerebellar ataxia have often been misdiagnosed with spinocerebellar degeneration, specifically multiple system atrophy with predominant cerebellar ataxia (MSA-C). However, more recently, it has been recognized that patients with PSP can present with truncal and limb ataxia as their initial symptom and/or main manifestation. These patients can be classified as having PSP with predominant cerebellar ataxia (PSP-C), a new subtype of PSP. Since the development of this classification, patients with PSP-C have been identified primarily in Asian countries, and it has been noted that this condition is very rare in Western communities. Furthermore, the clinical features of PSP-C have been identified, enabling it to be distinguished from other subtypes of PSP and MSA-C. In this review, we describe the clinical and neuropathological features of PSP-C. The hypothesized pathophysiology of cerebellar ataxia in PSP-C is also discussed.
In 1964, Steele et al. [1] defined the clinicopathological entities of progressive supranuclear palsy (PSP). They described PSP as a clinical disorder characterized by supranuclear gaze palsy, pseudobulbar palsy, dysarthria, dystonic rigidity of the neck and upper trunk and mild cognitive impairment. In addition, pathological examinations of PSP patients have revealed that subcortical regions such as the globus pallidus, subthalamic nucleus, substantia nigra, and cerebellar dentate nucleus are the most affected in this condition. Williams et al. [2] revealed that one-third of Caucasian patients with pathologically proven PSP develop parkinsonism, which is characterized by asymmetric onset, tremor, bradykinesia, and levodopa responsiveness. On the basis of this observation, two distinct clinical phenotypes of PSP—classic Richardson’s syndrome (PSP-RS) and PSP-parkinsonism (PSP-P)— have been proposed. Since then, several other clinical variants of PSP have been identified worldwide [3].
Although cerebellar ataxia is one of the exclusion criteria of the National Institute of Neurological Disorders and Stroke and the Society for Progressive Supranuclear Palsy (NINDS-SPSP) [4], it has been recognized that some patients with pathologically confirmed PSP present with truncal and limb ataxia as their initial symptom and/or main manifestation (Table 1). These individuals are often misdiagnosed with spinocerebellar degeneration (SCD), notably multiple system atrophy (MSA) [5] and idiopathic late onset cerebellar ataxias (ILOCAs) [6] or sporadic adult onset ataxia of unknown etiology (SAOA) [7]. This has led to the classification of a new subtype of PSP called PSP with predominant cerebellar ataxia (PSP-C) [5,8]. Studies into the pathology of PSP-C patients have demonstrated more severe neuronal loss and gliosis, higher densities of coiled bodies in the cerebellar dentate nucleus, and tau-positive granular profiles in Purkinje cells compared to PSP-RS patients [5,9]. These distinct features may give some indication of the underlying pathophysiology of cerebellar ataxia in PSP-C.
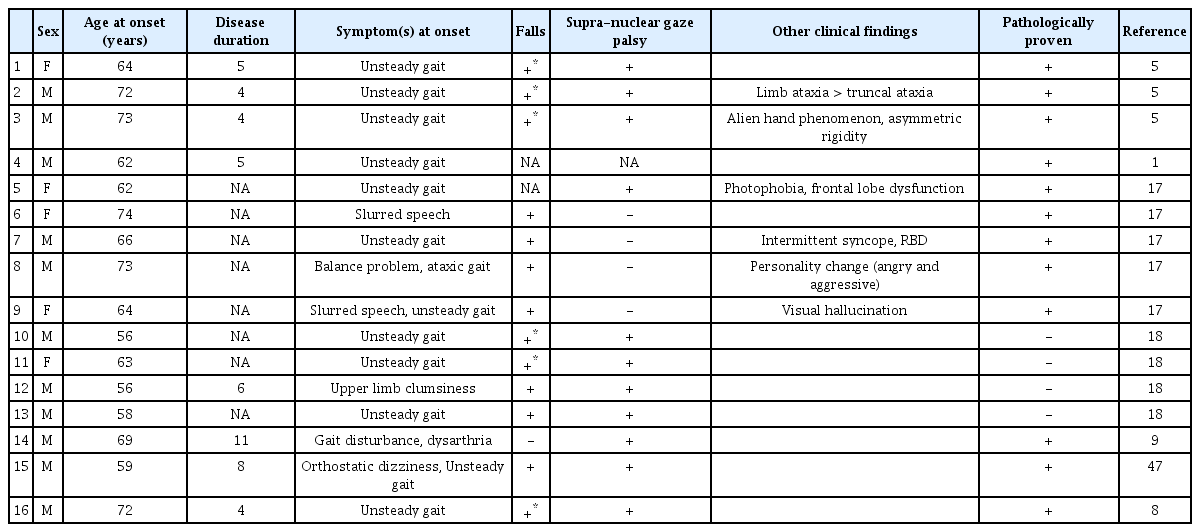
Case summary of PSP with predominant cerebellar ataxia
The aim of this review is to describe the clinicopathological characteristics of PSP-C. Furthermore, we discuss the pathophysiology of cerebellar ataxia and potential future investigations in PSP-C.
PSP SUBTYPES
It is important to understand the differences among PSP subtypes. First, we will briefly review the characteristics of PSP-RS and other PSP subtypes, excluding PSP-C.
PSP-RS accounts for approximately 50% of PSP cases [2,10] and is thus thought to be the most common PSP subtype. PSP-RS has established diagnostic criteria provided by NINDS-SPSP, which describe “probable PSP” as a gradual progressive disorder with an age at onset over 40 years, falls within the first year, and vertical supranuclear gaze palsy or slowing of vertical saccades [4]. In these NINDS-SPSP diagnostic criteria, cerebellar ataxia is one of the exclusion criteria [4]. In contrast, in the 2017 Movement Disorder Society Criteria, Höglinger et al. [11] suggest that prominent appendicular ataxia should be the only exclusion criterion. These diagnostic criteria do not include PSP-C as one of the recognized subtypes, given the rarity of this cerebellar ataxia predominant phenotype in the Western populations and the risk of misdiagnosing it as multiple system atrophy with predominant cerebellar ataxia (MSA-C) or ILOCAs [11]. The prognosis of PSP-RS is thought to be the worst among all PSP subtypes: the average disease duration is 5.9 years, and the average age at death is 72.1 years [12].
Patients with pathologically proven PSP have been observed to exhibit a wide variety of clinical presentations, leading to the subdivision of PSP phenotypes. PSP-P patients exhibit asymmetric limb bradykinesia and rigidity; hence, this condition is often misdiagnosed as idiopathic Parkinson’s disease (iPD). Although approximately half of PSP-P cases exhibit levodopa responsiveness, this honeymoon period is thought to be shorter than that of iPD. With respect to prognosis, the average survival time of PSP-P is nine years [12], thus PSP-P is typically more benign than PSP-RS [12]. Some additional PSP subtypes exhibit prominent clinical features: a freezing of gait and akinesia without axial and limb rigidity and vertical supranuclear gaze palsy (PSP-PAGF or PSP-PGF) [13] and a speech-exclusive impairment characterized by nonfluent aphasia with anarthria/apraxia of speech (PSP-AOS or PSP with predominant speech/language disorder, PSP-SL) [11,14].
CLINICOPATHOLOGICAL FEATURES OF PSP-C
Epidemiology
The prevalence of PSP-C exhibits significant regional differences. Cohorts of 30 European descendants in Australia [15] and 100 patients in a multicenter study in Europe and Canada [16] both failed to show any PSP-C patients, though the former cohort did include two patients presenting with cerebellar ataxia at an advanced stage. Furthermore, Koga et al. [17] demonstrated only one PSP-C patient among 100 cases of pathologically proven PSP. These researchers also calculated the frequency of PSP-C among 1,085 patients from the Brain Bank Mayo Clinic database, showing a low frequency of only 5 PSP-C patients (0.46%). On the other hand, three out of 22 pathologically confirmed PSP patients were reported as having “unclassified PSP” in a Japanese population. These individuals presented with cerebellar ataxia as their initial and principal manifestation, indicating a diagnosis of PSP-C [5]. Moreover, Xu et al. [18] demonstrated four clinically suspected PSP-C patients from China, and Iwasaki et al. [9] described the clinicopathological characteristics of a PSP-C patient from Japan. It should also be noted that one patient exhibited truncal and limb ataxia as their initial symptom and main manifestation, and three of nine patients exhibited truncal and/or limb ataxia during the disease course in the original paper first describing PSP (Table 1) [1]. In summary, it seems that PSP-C may be far more common in Asia than in Western nations, suggesting that genetic or environmental factors may play some role in the development of this phenotype.
Clinical findings
Clinical findings in PSP-C
Most patients with PSP-C present with gait disturbances associated with truncal ataxia as their initial symptom. The mean age at disease onset is 64.7 ± 6.5 years (range = 56–73). PSP-C predominantly affects men. All PSP-C patients exhibit falls, which occur at any stage of the disease. Some PSP-C patients may not exhibit supranuclear gaze palsy. The average disease duration from onset to death is estimated to be 6.0 ± 2.3 years.
With regard to nonmotor symptoms (NMSs), the frequency in each PSP subtype is not apparent. van Gerpen et al. [19] very recently reported that the prevalence of orthostatic hypotension and rapid eye movement (REM) sleep behavior disorder (RBD) in those with PSP (0% and 33%, respectively) was less than that in those with Lewy body disease (75% and 90%, respectively) and MSA (61% and 87%, respectively). As shown in Table 1, only one patient with PSP-C developed RBD and syncope (6%, 1/16), which suggests that there may be no differences in the frequency of NMSs in PSP-C and classical PSP. However, further studies are necessary to determine the differences.
Cognitive impairment in PSP-C seems to be similar to that in classical PSP. Koga et al. [17] demonstrated that the prevalence of memory cognitive complaints was not significantly different between the PSP-C group and other PSP groups. As PSP-C is a rare condition, a detailed profile of cognitive impairment, including frontal sign, aphasia, and personality change, has not been elucidated. Nevertheless, a PSP-C patient presented with frontal dominant cognitive dysfunction, and another PSP-C patient presented with visual hallucination (12%, 2/16; Table 1). This frequency seems to be compatible with the fact that 12% (12/100) of autopsy-proven PSP patients had developed frontal dysfunction [16].
Clinical differential diagnosis of PSP-C
PSP-C is often misdiagnosed as MSA-C. It is therefore crucial to understand the differences between these two diseases, particularly in their early stages [20]. Age at disease onset is higher in PSP-C than in MSA. Early falling is a distinctive feature of PSP-C, and this is uncommon in MSA. Moreover, supranuclear gaze palsy is common in PSP-C, whereas gaze-evoked nystagmus and abnormal eye pursuit movement are both characteristics of MSA.
As Höglinger et al. [11] mentioned, it is possible for PSP-C to be misdiagnosed as ILOCAs. However, based on the cohort study of PSP-C and ILOCAs, some clinical features may help to distinguish between them. Supranuclear gaze palsy may be more indicative of PSP-C because it has not been reported in either European or Japanese cohorts [6,7,21]. In addition, the incidence of cognitive impairment was less than 10% in ILOCAs; on the other hand, cognitive impairment has a higher prevalence in PSP-C (25% [5] to 80% [17]).
Regarding the radiological findings of PSP-C, no remarkable atrophy of the cerebellum or midbrain is observed during the very early phase of this disease [8]. However, the evaluation of serial MRI findings in a pathologically proven PSP-C patient revealed atrophy of the cerebellum and dilation of the pontocerebellar cistern, with no widening of the cerebellar fissures visible [8]. The observed atrophy and dilation were proportional to disease progression. Therefore, these MRI findings may be characteristic imaging features of PSP-C and help to differentiate between PSP-C and MSA-C or ILOCAs. Furthermore, atrophy of the midbrain tegmentum with the “hummingbird” sign was also observed in the advanced stage [22]. Longitudinally, atrophy of the midbrain tegmentum was common in PSP-C in the advanced stage. On the other hand, other MRI abnormalities, such as the “hot cross bun sign,” which is a distinctive feature of MSA, are not observed in PSP-C.
Neuropathology
In general, microscopic findings of PSP include neuronal loss, gliosis and the presence of neurofibrillary tangles (NFTs) in the basal ganglia and brainstem [23]. The most affected regions are the globus pallidus, subthalamic nucleus and substantia nigra. The subthalamic nucleus is thought to be particularly affected in the early phase, which may help to distinguish PSP from iPD. In terms of PSP-C, the fundamental pathological features are consistent with those of PSP. However, some unique neuropathological findings have been identified in PSP-C. For example, PSP patients presenting with cerebellar ataxia tend to exhibit greater neuronal loss and a higher number of tau-positive granular profiles in Purkinje cells [5]. In addition, more severe degeneration of the dentate nucleus is observed in PSP-C than in PSP [5]. Grumose degeneration in the dentate nucleus, indicative of the degeneration of terminal axons, is commonly observed [24,25]. However, grumose degeneration was not observed in the severe loss of Purkinje cells. Furthermore, Iwasaki et al. [26] also reported a PSP-C case with a prominent pathological change of the olivopontocerebellar system, including numerous glial fibrillary tangles and argyrophilic threads in the cerebellar cortex, deciduous Purkinje cells and tau-positive inclusions in the Bergmann glia and dentate nucleus neurons. It has therefore been described that cerebellar efferent pathways are commonly involved and suggested that these pathological changes may be associated with the pathophysiology of cerebellar ataxia in PSP-C. Conversely, Koga et al. [17] demonstrated that the severity of tau-related pathology within the cerebellar afferent pathway (for example, the inferior olivary nucleus) and other regions, such as the subthalamic nucleus and globus pallidus, did not differ between PSP-C and PSP-RS.
In terms of the difference between PSP-C and spinocerebellar ataxias (SCAs), SCA11 is especially important because this disease is caused by a tau tubulin kinase 2 (TTBK2) gene frameshift mutation [27], and the pathological change includes cerebellar atrophy with a severe loss of Purkinje cells and cerebellar granule cells [28]. Moreover, NFTs and tau-positive neurites can be observed in the midbrain tegmentum, substantia nigra, medullary tegmentum and putamen. It is notable that atrophy may be limited to the cerebellum. In contrast, in PSP-C, there is severe neuronal loss with gliosis in the dentate nucleus, and tau-positive granular profiles are present in the Purkinje cells; there is no severe loss of Purkinje cells and cerebellar granule cells. The presence of Purkinje cell and cerebellar granule cell loss might help to distinguish PSP-C and SCA11 pathologically.
It is also worth mentioning that PSP and MSA could coexist pathologically. Silveira-Moriyama et al. [29] reviewed several cases of coexistent PSP and MSA pathology. Neuronal loss was observed in the dentate nucleus, subthalamic nucleus, putamen and substantia nigra; however, some cases showed preserved dentate or subthalamic nuclei. In addition, tufted astrocytes/coiled bodies and glial cytoplasmic inclusions (GCIs) were observed simultaneously in these cases. The authors suggested that the coexistence of PSP and MSA could not be incidental because the prevalence of MSA pathology in patients with PSP (0.3% [30] to 0.8% [29]) was slightly higher than that in the general population (0.06% [31] to 0.4% [32]). To explain this phenomenon, it would be better to systematically screen the dual pathologies of PSP and MSA.
SUPPOSED PATHOPHYSIOLOGY OF CEREBELLAR ATAXIA IN PSP-C
The pathological changes that occur in PSP-C could spread throughout the cerebellum and involve Purkinje cells and the dentate nucleus. In particular, cerebellar efferent pathways, including the dentato-thalamo-cortical pathway, may be important to the origin of cerebellar ataxia in PSP-C (Figure 1). This potential theory is supported by the results of several clinical studies. First, the electrophysiological study of PSP patients showed a reduction in the motor evoked potentials (MEPs) elicited by transcranial magnetic stimulation (TMS) when they were preceded by stimulation from the cerebellum, which indicated cerebellar efferent pathway dysfunction [33]. Second, the involvement of the superior cerebellar peduncle (SCP), the cerebellar outflow tracts, was suggested. Several authors have shown atrophy [6] and increased fluid-attenuated inversion recovery (FLAIR) signal of SCP [34], respectively. Moreover, studies using novel MRI sequences, readout segmentation of long variable echo-trains (RESOLVE) [35] and diffusion tensor imaging (DTI) [36] demonstrated alterations in the decussation of SCP in patients with PSP, which implied pathological involvement of SCP in PSP. Third, given the predominant involvement of truncal ataxia rather than limb ataxia, the origin of cerebellar ataxia in PSP-C could be explained by a dentate lesion similar to that seen in dentatorubral pallidoluysian atrophy (DRPLA). This condition exhibits truncal-predominant ataxia, and it has been shown that there is significant degeneration of the dentate nucleus, without the involvement of the cerebellar cortex. In addition, patients with metronidazole-induced central nervous system toxicity exhibited marked postural instability and selective dentate nucleus lesions [37]. It is important to note that the deep cerebellar nuclei, including the dentate nucleus, communicate with Purkinje cells and the inferior olives and receive most of the cerebellar output information [38]. For example, Purkinje cells in the vermis are connected to the fastigial nucleus, one of the deep cerebellar nuclei, and project bilaterally to the reticular formation and lateral vestibular nuclei, which form the medial reticulospinal tract and the lateral vestibulospinal tract, respectively [39]. Moreover, PSP-C patients sometimes revealed palatal myoclonus due to the involvement of the dentate nucleus, red nucleus, and inferior olivary nucleus (the triangle of Guillain-Mollaret) [22]. These pathways are thought to be important for balance and postural control during voluntary motor tasks. Taken together, the evidence for the relationship between cerebellar ataxia in PSP-C and the cerebellar efferent pathway has been accumulating. However, most of the findings discussed above are not specific to PSP-C, and most PSP patients without cerebellar ataxia also have dentate pathological changes. Extrapyramidal symptoms and signs might mask the initial cerebellar signs [18]. The devices for the analysis of cerebellar function, such as stabilometry [4,40] and TMS, may help better understand cerebellar efferent pathway dysfunction in PSP-C.
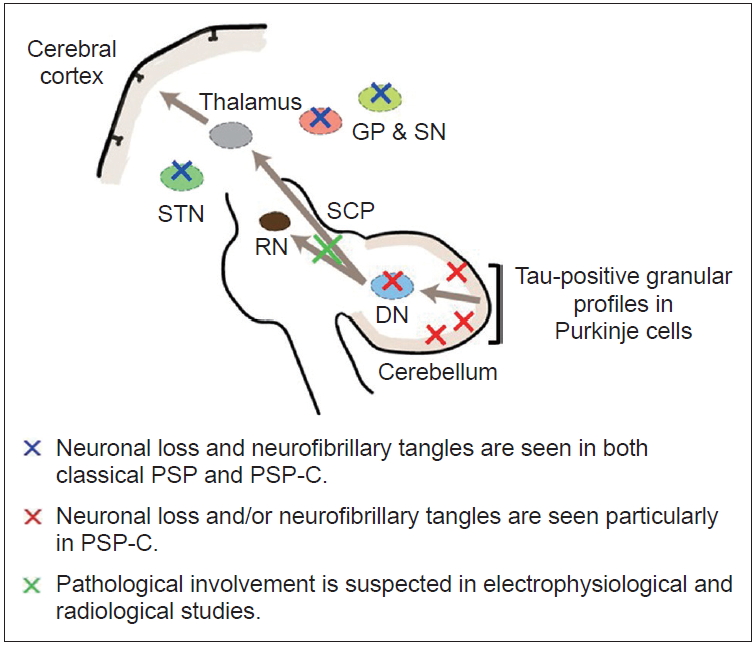
Schematic of the pathophysiology of cerebellar ataxia in predominant cerebellar ataxia (PSP-C). The arrows represent the cerebellar efferent pathway. DN: dentate nucleus, SCP: superior cerebellar peduncle, RN: red nucleus, STN: subthalamic nucleus, GP: globus pallidus, SN: substantia nigra.
THE UNKNOWN FIELD IN PSP-C TO BE CLARIFIED IN THE FUTURE
We demonstrated that early falls were significantly more frequent in PSP-C patients than in MSA-C patients in the two years following disease onset [20]. Conversely, the presence of cerebellar ataxia was similar in both PSP-C and MSA-C early in the disease course. It would therefore be interesting to understand the pathophysiology underlying these differences. Given that falls are cardinal features of not only PSP-C but also other PSP subtypes, it cannot be said that falls are caused by only cerebellar ataxia, which is characteristic for PSP-C. With respect to the backward and unprovoked falls seen in patients with early PSP, similar falls are recognized in patients with advanced iPD. In addition, pathological involvement of the subthalamic nuclei is cardinal in both PSP and advanced iPD. Falls are also frequently observed in iPD patients treated with deep brain stimulation of the subthalamic nuclei [41]. To compare pallidal versus subthalamic deep brain stimulation, falls are more frequent in the subthalamic group than in the pallidal group [42,43]. Subthalamic nuclei modulate basal ganglia circuits, and beta bursts in the subthalamic nuclei may affect motor performance [44]. Together, these facts lead us to consider that the subthalamic nuclei might be responsible for falls. Nevertheless, the circuit between the subthalamic nucleus and other basal ganglia and/or brainstem nuclei, including the red nucleus and inferior olivary nucleus, in falls and postural control remains unclear at this point. Moving forward, electrophysiological evaluation of the basal ganglia and/or brainstem nuclei may help to understand the physiology of motor performance in these aforementioned diseases.
With regard to the therapeutic intervention, PSP can be a candidate for disease-modifying therapies because PSP is a pure tauopathy. Thus, it is important to accurately diagnose PSP-C in the early phase for future investigations. Recently, Shimohata et al. [45] proposed certain diagnostic criteria for PSP-C. The required indications include (A) a slowly progressive course, (B) onset at age > 40 years, (C) supranuclear gaze palsy, (D) truncal and limb ataxia within 2 years of symptom onset, and (E) postural instability with falls within 2 years of symptom onset. Exclusion criteria include marked dysautonomia and the “hot cross bun sign” on a brain MRI. Probable PSP-C requires A + B + C + D + E, and possible PSP-C requires A + B + D + E. It should be cautioned that the criteria have some limitations. First, it is difficult to distinguish between PSP-C and ILOCAs and/or SAOA in the very early phases of the diseases. Second, validation studies of the diagnostic criteria for PSP-C have not been conducted. The sensitivity, specificity and accuracy of the diagnostic criteria are therefore unknown. However, in a study by Koga et al. [17] in 2016, according to these preliminary criteria, the symptoms of one patient were consistent with probable PSP-C, and the symptoms of 3 other patients were consistent with possible PSP-C. None of the 5 PSP-C patients had marked dysautonomia sufficient to meet Gilman’s criteria for a clinical diagnosis of probable MSA [46], and none had evidence of the hot cross bun sign on a brain MRI. The preliminary criteria for PSP-C seem to fit patients in the United States as well as those in Japan. A PSP-C patient developed orthostatic dizziness was reported, although dysautonomia was not pointed out [47]. Based on these considerations, it is desirable to evaluate more PSP-C patients to determine the specific findings for PSP-C and revise the current proposed criteria. Furthermore, studies to verify the diagnostic accuracy of PSP-C criteria should be conducted, particularly in Asian countries, which have a relatively high prevalence of PSP-C.
CONCLUSION
It has been recognized that patients with PSP can present with truncal and limb ataxia as their initial symptom and/or main manifestation. These patients can be grouped into the new classification called PSP-C. The accumulation of knowledge in PSP-C has suggested a difference in clinicopathological features between PSP-C and MSA or SCD. Moreover, clinical diagnostic criteria for PSP-C have been proposed. Further studies are needed to establish PSP-C as one of the PSP subtypes.
Notes
Conflicts of Interest
The authors have no potential conflicts of interest.
Author Contributions
Conceptualization: Masato Kanazawa. Data curation: Shoichiro Ando and Masato Kanazawa. Formal analysis: Shoichiro Ando and Masato Kanazawa. Funding acquisition: Masato Kanazawa. Investigation: Shoichiro Ando and Masato Kanazawa. Methodology: Shoichiro Ando and Masato Kanazawa. Resources: Shoichiro Ando and Masato Kanazawa. Software: Shoichiro Ando and Masato Kanazawa. Supervision: Osamu Onodera. Validation: Masato Kanazawa. Visualization: Shoichiro Ando. Writing-original draft: Shoichiro Ando. Writing-review & editing: Masato Kanazawa and Osamu Onodera.
Acknowledgements
The PSP-C phenotype has become well recognized due to efforts by Professor Shimohata (Gifu University, Japan) and helpful suggestions from Honorary Professor Nishizawa (Niigata University, Japan). We thank Dr. Y. Toyoshima, Dr. M. Tada, Professor A. Kakita, and Professor H. Takahashi (Niigata University, Japan) for their contributions. We also thank our predecessors, especially Professor Steele, Professor Richardson, and Professor Olszewski, who provided valuable detailed clinical records.