Immunotherapy Targeting Neurodegenerative Proteinopathies: α-Synucleinopathies and Tauopathies
Article information

Abstract
α-Synuclein and tau deposition in the central nervous system is responsible for various parkinsonian syndromes, including Parkinson’s disease, multiple system atrophy, dementia with Lewy bodies, progressive supranuclear palsy and corticobasal degeneration. Emerging evidence has suggested that pathologic α-synuclein and tau are transmitted from cell to cell and further accelerate the aggregation of pathologic proteins in neighboring cells. Furthermore, extracellular pathologic proteins have also been reported to provoke inflammatory responses that lead to neurodegeneration. Therefore, immunotherapies targeting extracellular α-synuclein and tau have been proposed as potential disease-modifying strategies. In this review, we summarize completed phase I trials and ongoing phase II trials of immunotherapies against α-synuclein and tau and further discuss concerns and hurdles to overcome in the future.
Most neurodegenerative diseases involve characteristic pathologic protein aggregation in the central nervous system with a loss of neurons in characteristic anatomical locations [1]. The neuropathological criteria of Parkinson’s disease (PD) consist of two histopathological features: the loss of dopaminergic neurons and α-synuclein deposition in the substantia nigra [2]. Subsequent studies have found that a series of parkinsonian disorders with various progression rates and distinct clinical features, such as multiple system atrophy (MSA) and dementia with Lewy bodies (DLB), share common pathologic features. Although the dominantly affected cell types and anatomical locations differ among the disorders, a common feature is the aggregation of α-synuclein in the central nervous system [3]. Additionally, distinct atypical parkinsonian syndromes, such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), share the common neuropathologic feature of tau deposition in the brain [4]. Therefore, substantial research has been conducted to elucidate the role of α-synuclein and tau as the pathogenic causes of neurodegenerative diseases.
Intraneuronal inclusions of aggregated proteins in PD were first reported in 1912 and were named Lewy bodies [5]. In 1997, aggregated α-synuclein was identified in Lewy bodies [6], and this raised the question of whether aggregated α-synuclein is the cause of the disease or exists as bystander or end product of the disease. Based on clinical and preclinical studies from the past two decades, there is evidence that α-synuclein is the cause and driver of the disease. The first evidence came from genetic analysis of familial cases. In 1997, missense mutations in the SNCA gene, which encodes synuclein, were found in patients with early-onset autosomal dominant familial parkinsonism [7]. Furthermore, familial parkinsonian patients harboring duplication/triplication of the SNCA gene were reported, and the fact that patients with triplications showed much more severe symptoms suggested that the burden of α-synuclein might be related to the disease [8,9]. The next evidence came from neuropathologic studies showing an association between α-synuclein pathology and disease progression. Based on the Braak hypothesis, α-synuclein depositions start in the olfactory bulb or dorsal motor nucleus of vagus, which is connected to the enteric nervous system via the vagus nerve. Deposition further spreads to the midbrain, basal ganglia and cortex and correlates with the clinical progression of the disease [10]. Furthermore, a large number of in vivo and in vitro animal studies have shown that α-synuclein, especially in the oligomeric form, is toxic to neurons and leads to neurodegeneration [11-13]. Additionally, it is well known that phosphorylated tau or acetylated tau plays a critical role in toxicity in the central nervous system, which leads to neurodegeneration [14]. Similar to synucleinopathies, soluble tau oligomers are considered the most toxic form of tau [15].
Notably, there is no curative or disease-modifying treatment available for synucleinopathies or tauopathies. There are no therapeutic strategies targeting pathologic protein aggregation in the brain. Therefore, it is reasonable and important to target pathologic proteins such as the oligomeric form of α-synuclein or tau as potential disease-modifying strategies. Currently, there are many strategies targeting pathologic proteins, such as those that increase clearance [16-18] and posttranslational modifications [19,20] and inhibit aggregation [21-23]. In this review, we will focus on immunotherapies targeting α-synuclein and tau.
STRUCTURE AND FUNCTION OF α-SYNUCLEIN
α-Synuclein is a member of the synuclein family (which includes α, β, γ-synuclein and synoretin) and is translated from the SNCA gene located on chromosome 4q21-23 [7]. Synuclein was first discovered in cholinergic neurons in Torpedo californica (the Pacific electric ray) as a protein localized to synaptic vesicles and nuclei [24]. The name synuclein came from its localization to synapses and nuclei. α-Synuclein is highly expressed in the brain, especially in presynaptic terminals of neurons, as well as red blood cells and platelets [25,26]. Little is known about the physiologic role of native α-synuclein. However, studies have suggested that it is closely related to the regulation of synaptic vesicle dynamics [27]. It closely interacts with the SNARE (Soluble N-Ethylmaleimidesensitive factor Attachment protein Receptor) complex, which plays a critical role in the release of neurotransmitters [28,29]. Furthermore, there have been studies suggesting that α-synuclein plays a role in striatal dopamine release [30]. Its primary sequence is composed of 140 amino acids and contains 3 main domains: the N-terminal domain, C-terminal domain and non-amyloid-component (NAC) domain (Figure 1) [31]. The N-terminal domain is a highly conserved lysine-rich zone [32]. All previously reported mutations are in the N-terminal domain area, which shows that this region plays a critical role in aggregation. The NAC domain is a hydrophobic domain that enables α-synuclein to aggregate into β-sheet-rich fibrils. In the C-terminal region, the majority of posttranslational modifications, such as phosphorylation at serine 129, occurs [33,34], and truncation of this area is related to an increased rate of α- synuclein aggregation [35].
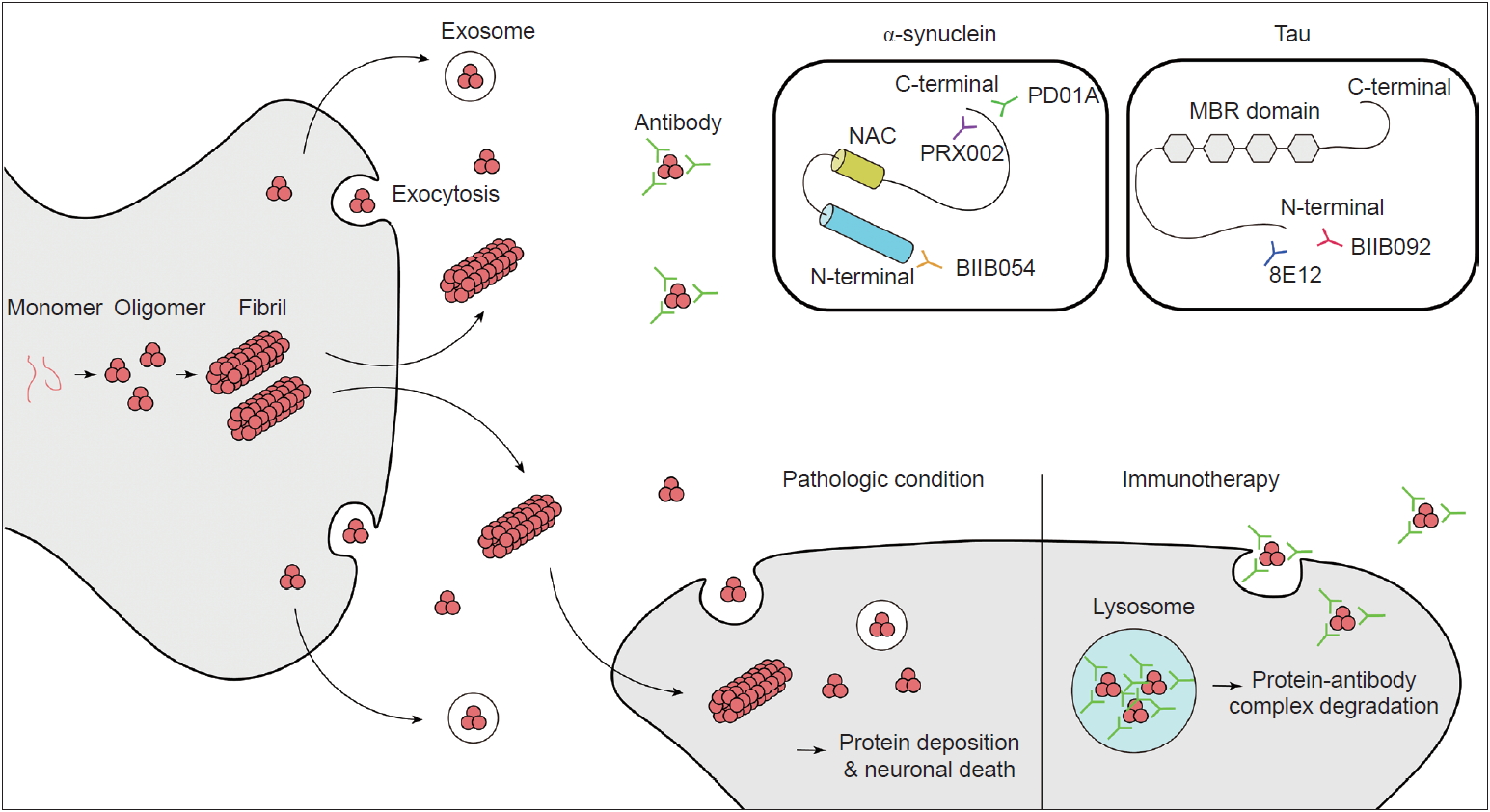
Schematic illustration of cell-to cell propagation of α-synuclein and Tau with mechanism of immunotherapy. Binding sites for antibodies which is on current clinical trials are presented. NAC: non-amyloid component, MBR: microtubule-binding repeats.
There has been some debate about the structure of the native state of α-synuclein. It was previously thought to be unstructured in the native state; however, some studies have shown that it exists as a helically folded tetramer [36]. Under physiologically stressed conditions such as high temperature or low pH, α-synuclein adopts a partially folded intermediate structure [37]. As the partially folded structure contains hydrophobic patches on its surface, it is likely to be aggregated to form beta-sheet structures and form amyloid fibrils. As monomeric α-synuclein lacks hydrophobic patches, conditions that mediate the formation of the partially folded intermediate form are considered pathogenic [38]. The mechanism of the initial conformational change from normal monomeric α-synuclein to the pathologic form is largely unknown. However, it has been reported that fibril formation is accelerated in the presence of familial PD-associated α-synuclein mutations (E46K, A53T and H50Q), although some mutations (A30P, G51D and A53E) are associated with decreased fibril formation rates in vitro [39]. There are two phases of α-synuclein aggregation. One is primary nucleation, which is the formation of oligomers and fibrils from monomers; the other is the secondary nucleation phase, which is the elongation of fibrils or the transformation of oligomers from monomers by surface-catalyzed reaction by fibrils [39].
TOXICITY OF α-SYNUCLEIN AND ITS CELL-TO-CELL PROPAGATION
Other important questions are concern the form of α-synuclein that is toxic to neurons and the mechanism of this toxicity. Originally, it was thought that mature inclusion bodies cause neurodegeneration, but this has come under debate. Accumulating evidence has shown that it is the oligomeric intermediate that is toxic and causes neurodegeneration [40-42]. The main mechanism has been suggested to be increased membrane permeability, which leads to disruption of ionic homeostasis, depolarization of the mitochondrial membrane, and an increase in reactive oxygen species followed by caspase3 activation [43]. Additionally, the proposed mechanisms of α-synuclein toxicity include mitochondrial dysfunction [44-46], disruption of endoplasmic reticulum and Golgi trafficking [47,48], defective autophagy [49], disruption of interorganelle contacts [50] and altered transcription factor activation [51,52]. One of the amazing features of α-synuclein is that it can propagate from cell to cell (Figure 1). This was suggested by studies that demonstrated Lewy-body-like inclusions in grafted embryonic nigral neurons, which led to the hypothesis of the host-to-graft transmission of α-synuclein [53,54]. Similarly, in Braak’s autopsy study, it was hypothesized that α-synuclein aggregates spread to interconnected neural structures [10]. This observation cannot be readily explained if α-synuclein depositions occur spontaneously in each neuron. Subsequent studies have found that a portion of α-synuclein is present in vesicles in the cytoplasm and is secreted by exocytosis (Figure 1) [55]. Extracellular α-synuclein can exist as oligomers or fibrils, which can be internalized by neighboring neurons and undergo endosomal trafficking followed by lysosomal degradation [56,57]. If a fibril survives lysosomal degradation, it can accelerate α-synuclein aggregation through the mechanism mentioned above (secondary nuclearization). In addition, extracellular α-synuclein acts on the TLR2 signaling pathway in glial cells and produces an inflammatory response, which also plays a role in neurodegeneration [58]. Therefore, extracellular α-synuclein is currently considered a major pathogenic process of and a novel treatment target for synucleinopathies.
IMMUNOTHERAPY INVOLVING α-SYNUCLEIN
Many antibodies have been studied and proposed as potential blockers of α-synuclein aggregation and propagation. The expected mechanisms are: 1) blocking extracellular α-synuclein in the brain to reduce cell-to-cell transmission, 2) reducing the total burden of the pathologic forms (oligomers/fibrils) of α-synuclein while sparing the physiologic forms (monomers/tetramers) of α-synuclein, and 3) stopping or slowing disease progression and hopefully reversing the symptoms of PD.
There are two main ways of delivering antibodies against α-synuclein. One is passive immunization, and the other is active immunization. Several antibodies and vaccines (peptides used to induce an immune response to produce antibodies against α-synuclein) targeting the C-terminus or N-terminus of synuclein or synuclein oligomers have been studied in preclinical studies; these studies showed decreased neuronal loss with behavioral benefits in PD animal models [59-63]. Among them, several antibodies have been tested in human clinical trials. As of April 2019, phase I trials have been completed for two passive immunization antibodies and one active immunization peptide, and phase II trials are ongoing (Table 1 and 2).

Completed phase I trials of α-synuclein-targeted immunotherapy

On-going phase I/II trials of α-synuclein- and tau-targeted immunotherapy in Parkinsonian syndromes
PRX002
PRX002, also known as the 9E4 antibody, was the first therapeutic antibody developed by Prothena. The antibody targets the C-terminus of α-synuclein (Figure 1) and has more than 400-fold greater affinity for oligomeric α-synuclein than for monomeric α-synuclein. A preclinical animal model study showed that the passive delivery of the antibody blocked cell transmission, lowered intracellular α-synuclein pathology, and restored motor and cognitive deficits in multiple transgenic mouse models of PD [59,63]. Important findings were reported after the completion of the phase I trial [64]. The results showed that, at a dose of up to 60 mg/kg, it was tolerable and safe in PD patients. The serum levels of PRX002 were increased in a dose-dependent manner, and the serum α-synuclein level was reduced by up to 97% at the highest dose (60 mg/kg) [64]. The cerebrospinal fluid (CSF) level of PRX002 also increased proportionally with the dose. However, the ratio of the CSF PRX002 level and the serum PRX002 level was approximately 0.3% across all doses [64]. Interestingly, there was no change in the CSF α-synuclein level [64]. PRX002 is currently in a phase II clinical trial (PASADENA study, clinical trial identifier NCT03100149).
BIIB054
BIIB054 is a human anti-α-syn IgG1 monoclonal antibody also known as the 12F4 antibody. This antibody targets the N-terminus of α-synuclein (Figure 1). It is very selective for the aggregated form of α-synuclein (more than 800-fold) [65]. BIIB054 also showed good safety and tolerance at single doses up to 135 mg/ kg in healthy controls and 45 mg/kg in PD patients [65]. BIIB054 displayed dose-proportional pharmacokinetics and dose-dependently induced the formation of a BIIB054/α-synuclein complex in the serum [65]. The CSF-to-serum ratio of the antibody was between 0.3% (45 mg/kg group) and 0.5% (15 mg/kg group) when measured 4 weeks after injection in PD patients [65]. However, whether there was a significant decrease in the CSF α-synuclein level was not reported [65]. Currently, BIIB054 is being evaluated in a phase II study in early PD patients (SPARK study, clinical trial identifier NCT03318523).
PD01A/PD03A
Currently, there is only one company (AFFiRis, Wien, Austria) that has developed active immunization (a vaccine) targeting α-synuclein. α-Synuclein AFFITOPE (AFF) is a short peptide that mimics the α-synuclein molecule but has an amino acid sequence that is different from that of the native protein. It was designed to induce a B cell immunogenic response to produce an antibody without inducing a T cell-related autoimmune response [66]. PD01A and PD03A induced the robust production of antibodies that selectively recognize aggregated α-synuclein while sparing other synuclein family members and monomeric α-synuclein [66]. The peptide has been tested in animal models of synucleinopathies, including PD, MSA and DLB [66,67]. In animal models of synucleinopathies, vaccination reduced the burden of aggregated α-synuclein and neurodegeneration. Furthermore, it rescued the motor functions of PD and MSA model mice [66,67]. Most importantly, AFF did not induce any neuroinflammation or neural damage. AFF has undergone phase I clinical trials, including in both early PD and healthy controls (clinical trial identifier NCT01568099). Four injections of 15 µg or 75 µg of AFF peptide with aluminum oxyhydroxide adjuvant were administered every 4 weeks. The results showed a good safety profile without serious adverse events, and the participants were followed up for a longer period (52 weeks, clinical trial identifier NCT01885494). After successful phase I trials, participants underwent boost immunization with the peptide and showed clear and robust induction of α-synuclein antibodies after the first boost, and the second boost further stabilized the antibody titers [68]. PD01A-induced antibodies preferentially bind to both the oligomeric and fibril forms of α-synuclein rather than monomers. Furthermore, there was a trend for α-synuclein levels in the plasma and CSF to be reduced at week 26. However, the clinical scores of the vaccine-treated group did not show any difference compared with those of the untreated control group after 12 months of observation. Subsequent phase I studies have targeted both early PD (PD03) and early MSA patients (PD01/PD03).
IMMUNOTHERAPY AGAINST TAU
Tauopathies are a series of neurodegenerative disorders with various clinical and pathological presentations characterized by the aggregation of abnormal tau proteins. Tau plays a role in the polymerization and assembly of microtubules in the normal physiologic state [69]; therefore, tau is found in the axons of neurons and in glial cells, including astrocytes and oligodendrocytes [70]. There are 6 isoforms of tau, which are classified based on the number of microtubule binding-potential repeat domains (3R or 4R) and the number of amino-terminal inserts (0N, 1N or 2N) [71,72]. Phosphorylated or acetylated tau creates filamentous aggregates in neurons or glial cells, causing neurodegeneration. Tau normally exists in an equal ratio of 3R:4R tau in nondisease states, and tauopathies can be classified based on the ratio of 3R:4R tau [4]. Atypical parkinsonian syndromes, including PSP and CBD, are related to 4R dominant tauopathies [2,4]. Based on promising results from preclinical trials [73-75], anti-tau antibodies have been tested in clinical trials in patients with PSP or CBD. As of April 2019, there are three passive immunization tau antibodies (BIIB092, C2N-8E12 and UCB0107) for ongoing clinical trials (Table 2, 3). BIIB092 is a humanized IgG4 monoclonal anti-tau antibody developed by Biogen. A phase I trial of BIIB092 was completed in patients with PSP, and BIIB902 was reported to be safe in a multiple ascending dose study (clinical trial identifier NCT02460094). The antibody showed a dose-dependent accumulation in the serum and CSF [76]. There was a marked reduction in the CSF level of free eTau, which is an N-terminal tau fragment known to be related to the progression of the disease (Figure 1) [76]. The reduction rate was over 90% in all doses, and this reduction was maintained over 85 days [76]. BIIB092 is currently in a phase II trial aiming to recruit 396 patients with PSP (clinical trial identifier NCT03068468, PASSPORT). In addition, BIIB092 is currently in a phase Ib trial including CBD patients (clinical trial identifier NCT03658135, TauBasket). The 8E12 antibody is also a humanized IgG4 antibody developed by C2N Diagnostic and AbbVie. A phase I trial of the 8E12 antibody was completed (clinical trials identifier NCT02494024). The antibody has a half-life of 27 to 37 days, and a dose-dependent increase in the serum level was shown [77]. The CSF-to-serum ratio of the antibody was 0.18 to 0.35 [77]. The participants experienced no serious treatment-related adverse events [77]. Currently, the antibody is in phase II trials in PSP patients (clinical trials identifier NCT03391765).
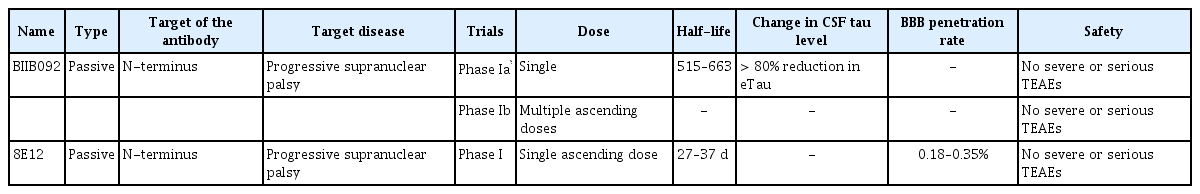
Completed phase I trials of tau-targeted immunotherapy
FUTURE DIRECTIONS
There are some active clinical trials of therapies targeting α-synuclein and tau in parkinsonian syndromes, including PD, MSA, PSP and CBD. Based on all phase I trials of α-synuclein immunotherapies, antibodies and peptides show good tolerability without severe adverse events. Additionally, studies have consistently shown in a time- and dose-dependent manner. However, there may be some concerns. Phase I studies have shown a reduction in the serum α-synuclein concentration upon the formation of antibody/α-synuclein complexes. However, no study has shown a significant reduction in CSF α-synuclein levels. Furthermore, the ratio of the CSF concentration to the serum concentration of the antibody was less than 0.5% in passive immunization trials. The authors claimed that this was because a large portion of α-synuclein in the CSF is monomeric and that the PRX002 concentration in the CSF was not high enough to engage monomers, as PRX002 has a higher affinity (> 400-fold) for oligomers [64]. However, it remains unclear whether the CSF level of the antibody is sufficient with the currently used peripherally (intravenously) injected dose. The CSF penetration rate of other IgG monoclonal antibodies, including in preclinical animal studies, was reported to range from 0.1–1% [78-80]. Thus, determining optimal CSF antibody concentrations that can alter α-synuclein and tau propagation and deposition in humans would help establish an optimal peripheral (intravenous) dose of the antibody; this would require further studies regarding biomarkers of pathologic burden. On the other hand, we may have to consider alternative strategies, including direct intrathecal injection or increasing BBB penetration. Recently developed focused ultrasonography has been widely used to ablate the thalamus and STN in ET and PD patients [81,82]. One of the collateral damages is that it opens the local blood-brain barrier [83]. As this is a safe, noninvasive procedure, it might be considered an adjuvant strategy for peripheral passive immunization.
The next concern relates to the efficacy of immunization. In AD, the amyloid beta-targeting antibodies bapineuzumab and solanezumab failed to stop cognitive decline after 72 weeks and 78 weeks, respectively [84,85]. A recently phase III trial of aducanumab, which is also an anti-amyloid beta antibody, was halted. Possible explanations for these results include that amyloid beta is an ineffective target compared with tau and that the participants in the trials exhibited disease that was too advanced for recovery. Considering the lessons learned from amyloid beta immunization trials in AD, there might be important points to consider.
First, clinical trials should enroll patients in the early stage of disease. However, there are several hurdles to overcome. The diagnosis of PD, MSA and DLB is based on clinical evaluation, and there is a relatively high misdiagnosis rate (18–25%) [86-88]; he misdiagnosis rate tis even higher in patients in the early stage of the disease (35–47%) [89,90], who are suitable candidates for disease modifying treatment. There is a lack of biomarkers for early diagnosis and for monitoring the severity of the disease, which makes early diagnosis more difficult. Second, PD may manifest as a neurodegenerative disorder with mixed pathology that includes α-synuclein, tau and amyloid beta [91-93]. Therefore, clinically diagnosed and enrolled PD patients may be heterogeneous patients with different combinations of proteinopathies. Before considering immunotherapies targeting α-synuclein, it may be important to consider whether blocking and reducing the burden of α-synuclein alone is enough to ameliorate motor and nonmotor symptoms. Evaluating the correlation between clinical stage and the burden of α-synuclein aggregation, if possible, would lead to better targeted treatment strategies. In other words, without reliable biomarkers that reflect central α-synucleinopathy and the clinical stage, clinical trials of immunizations may repeatedly face similar limitations.
CONCLUSION
Immunotherapies against α-synuclein and tau are promising disease-modifying treatment strategies with acceptable safety profiles. However, there are several hurdles to overcome for future clinical trials. These challenges include optimizing the delivery of antibodies to the brain, recruiting patients in the early stage of the disease and developing biomarkers that reflect central α-synuclein/ tau pathology.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Author Contributions
Conceptualization: Junghwan Shin and Han-Joon Kim. Visualization: Junghwan Shin. Writing—original draft: Junghwan Shin. Writing—review & editing: All authors.