Modeling α-Synuclein Propagation with Preformed Fibril Injections
Article information

Abstract
The aggregation of α-synuclein (α-syn) has been implicated in the pathogenesis of many neurodegenerative disorders, including Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA). Postmortem analyses of α-syn pathology, especially that of PD, have suggested that aggregates progressively spread from a few discrete locations to wider brain regions. The neuron-to-neuron propagation of α-syn has been suggested to be the underlying mechanism by which aggregates spread throughout the brain. Many cellular and animal models has been created to study cell-to-cell propagation. Recently, it has been shown that a single injection of preformed fibrils (PFFs) made of recombinant α-syn proteins into various tissues and organs of many different animal species results in widespread α-syn pathology in the central nervous system (CNS). These PFF models have been extensively used to study the mechanism by which aggregates spread throughout the brain. Here, we review what we have learned from PFF models, describe the nature of PFFs and the neuropathological features, neurophysiological characteristics, and behavioral outcomes of the models.
α-Synuclein (α-syn) is a 140-amino acid protein in the synuclein family, which also includes β- and γ-synuclein [1,2]. Although the function of α-syn remains elusive, studies have suggested it has roles in synaptic transmission [3,4], lipid metabolism and transfer [5], and protein folding [6,7]. The most extensively studied aspect of the protein is its role in the pathogenesis of Parkinson’s disease (PD) and related disorders, including dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), which are collectively referred to as synucleinopathies [8,9]. In 1997, Polymeropoulos et al. [10] reported the first link between α-syn and early-onset PD. Subsequent genetic studies have also confirmed the link between both familial and sporadic PD and α-syn [11-14].
Synucleinopathies are a group of human neurodegenerative diseases characterized by the accumulation of α-syn in various regions of the central and peripheral nervous systems [15], and PD is the most common disease. Numerous results have pinpointed the misfolding and aggregation of α-syn as the culprit of synucleinopathies [14]. The abnormal aggregates, the nature of which remains elusive, can trigger downstream cascades, leading to neuroinflammation and neurodegeneration [16]. In PD, it has been suggested that α-syn aggregates spread from a few discrete regions to wider brain areas as the disease progresses [17]. In recent years, a growing body of evidence has suggested that the cell-to-cell propagation of α-syn is the underlying principle of aggregate spreading [18,19]. According to this “propagation hypothesis”, the spreading of aggregates is perpetuated as protein seeds are transferred from donor cells to recipient cells. With this ability to self-propagate, a small amount of preformed fibrils is sufficient to induce the conversion of normal proteins into insoluble aggregates, an event that can spread throughout the brain in a sequential manner via neural connections [20,21].
A limitation of postmortem analyses in humans is that they only portray the late stages of the disease, making it difficult to understand the initiation and progression events. The major obstacle of the field is to create animal models that can recapitulate the hallmark features and disease principles of human disease. Numerous transgenic (Tg) models have been developed and analyzed and dominate the field. Now, with the advent of the propagation hypothesis, new models that recapitulate the progression of protein aggregate pathology provide us opportunities to investigate how the disease progresses [22].
ANIMAL MODELS OF α-SYN AGGREGATE PROPAGATION
A variety of animal models have been developed for α-syn propagation research, and these models can be classified into three main types based on the approach used: 1) cell grafts into Tg mice expressing human α-syn, 2) α-syn overexpression by viral vectors and 3) the injection of in vitro-generated α-syn aggregates [22].
The cell graft model was inspired by the findings that Lewy body pathology is present in embryonic neurons transplanted into the striatum of PD patients [23,24]. In this model, green fluorescent protein (GFP)-labeled mouse neural precursor cells were injected into the brain regions of Tg mice expressing human α-syn [25]. The transfer of host-expressed human α-syn to grafted cells can be monitored by using human-specific α-syn antibodies.
The viral model was inspired by the hypothesis that α-syn pathology initiates in the enteric nervous system and enters the CNS through migration along the vagus nerve [26]. An adeno-associated virus (AAV) carrying human α-syn cDNA was injected into the vagus nerve of naïve rats or mice, and the transfer of human α-syn from the dorsal motor nucleus of the vagus through the pons and the midbrain to the frontal cortex was assessed by immunohistology [26].
Finally, there are various injection models in which exogenous α-syn preparations have been delivered to the brain. In these models, either brain extracts containing α-Syn aggregates (from Tg mice or patients with α-syn pathology) or in vitro-generated PFFs were injected into the mouse brains. The injected mice developed Lewy body-like pathology with enhanced phospho-α-syn (p-α-Syn) staining, neuronal loss, and some behavioral deficits. Such injections have been administered not only in the brain but also in many other areas in the body, including the muscle [27], gut [28], olfactory bulb [29], and peritoneum [30] (Figure 1). The PFF model is the most extensively used model for the study of α-Syn propagation, and we will review what has been documented and what can be learned with this model.
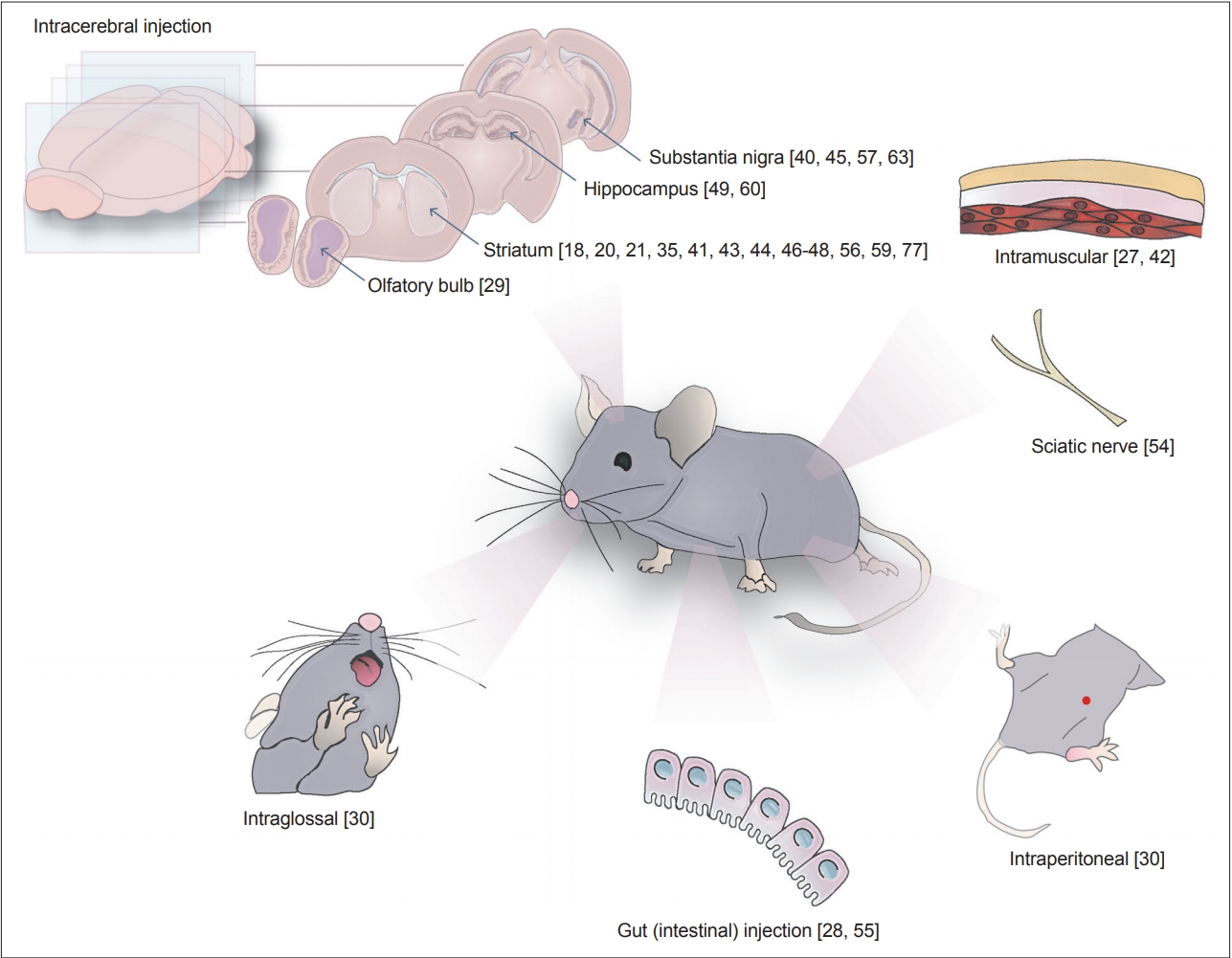
Sites used for preformed fibril inoculation. α-synuclein aggregates have been delivered to different sites through intracranial, gastrointestinal, intramuscular and intraglossal routes. The numbers correspond to the reference numbers of the original articles that performed the injections on the sites.
PREPARATION AND CHARACTERISTICS OF PFFS
PFFs are generated from monomeric recombinant α-syn proteins incubated under defined conditions [20,31]. These fibrils are then sonicated to form short fragments that can trigger the conversion of normal endogenous α-syn into the pathological form [18]. The introduction of PFFs in vitro or in vivo can induce endogenous α-syn to be hyperphosphorylated at Ser129 and ubiquitinated and become detergent-insoluble [19,20]. These changes in protein structure are thought to subsequently trigger a series of events, such as neuronal cell loss and behavioral deficits [18].
Validations of homemade or purchased monomeric α-syn proteins are crucial since not all α-syn monomers can aggregate and not all α-syn aggregates induce pathology [31]. A newly established protocol from the Michael J Fox Foundation for Parkinson’s Research involves 3 stages for the proper preparation of α-syn PFFs from monomers: 1) the preparation of the monomers, 2) the generation of PFFs from the monomers, and 3) the preparation of PFFs for experiments [32]. In brief, monomeric α-syn that is suitable for PFF development is centrifuged, the supernatant is transferred, and the protein concentration is measured. A portion of the starting monomers is retained for use as a negative control for quality control experiments of fibrils. To generate PFFs, the protein solution is diluted to 5 mg/mL, vortexed, incubated at 37°C with shaking for 7 days, and subjected to a quality test. α-Syn PFFs should not be stored at 4°C or -20°C because dissociation might occur. They should only be stored at -80°C [33]. It is advisable to use freshly prepared PFFs. Samples stored in the shortterm at room temperature or at -80°C for up to 1.5 years retain pathogenicity but can lose activity [34]. To generate pathogenic species of PFFs, the fibrils should be diluted and sonicated immediately before use. To achieve consistent pathogenicity, sonication must produce short fibrils with a length of 50 nm or shorter as the pathogenicity of α-syn PFFs depends on the structure and size of the fibrils [35,36]. Other than regular quality control experiments used to validate the conversion of monomeric α-syn to PFFs, in vivo pilot studies are required to validate the pathogenicity of PFF species before any long-term in vivo studies. All parameters at every step in the standard protocol should be validated and tailored to each study’s purposes and models [34].
The PFF model presents some advantages over previous disease models. In the PFF model, small seeds of nonphosphorylated PFFs induce the transformation of endogenously expressed α-syn into pathological aggregates, while other models are based on the overexpression of human WT or mutant α-syn [31,37]. Therefore, the levels of α-syn are more physiologic in the PFF model than in viral vector-based and Tg models. The time course of PFF-induced degeneration with early α-syn pathology in PD-related brain areas is similar to that in human conditions, in which dopamine neuron dysfunction precedes overt motor symptoms [38]. The model also enables us to follow the progression of α-syn aggregates throughout the course of their propagation, from early formation to the point of neuron death. Because of these features, the PFF model has great temporal and spatial resolution [31].
Nevertheless, experts have suggested some constraints that need to be considered when employing the PFF model. Since it is well established that unilateral injections of PFFs can produce bilateral pathology, it is not possible to use the contralateral side as an internal control [18,20,39]. Moreover, it is a great challenge to maintain the reproducibility of studies using the PFF model considering that the experiments often take months to complete [34]. The variations in the implementation of the experiments, including monomer preparation, the generation of PFF seeds, and the amount of injected PFFs, can also affect the consistency of the results.
SYNUCLEINOPATHIES IN INTRACEREBRAL PFF INJECTION MODELS
The distribution of α-syn inclusions after PFF injection is wide and diverse depending on the injection site, the amount and type of PFFs, and the animal species/lines. Luk et al. [20] demonstrated that injecting brain homogenates from M83 mice overexpressing human A53T α-syn into both the somatosensory cortex and dorsal neostriatum of asymptomatic α-Syn Tg mice induced the earlier onset and progressive spreading of Lewy body-like pathology. Immunohistochemistry results of p-α-syn as a pathological α-syn marker show abundant α-syn inclusions throughout CNS pathways, including the frontal cortex, thalamus, hypothalamus, brainstem nuclei, and major white matter tracts, 90 days postinjection [20]. The injection of both brain homogenates from Tg mice and synthetic PFFs, even into naïve mice, accelerates the formation of pathological Lewy bodies and Lewy neurite-like inclusions and induces propagation throughout the CNS to regions far away from injection sites [18,20,21,35,40-42]. A single intrastriatal injection of recombinant mouse α-syn PFFs [18,43,44] and of human α-syn PFFs [21,41,44] results in widespread α-syn pathology 90 and 180 days postinjection in WT mice.
At an early stage (30 days) after the injection of PFFs assembled from human α-syn1-120Myc or WT full-length human α-syn into the striatum and cortex, p-α-syn pathology can be detected mostly in sites connected to the injection site, including the ventral striatum, thalamus, occipital cortex, and brainstem fibers [20]. These results suggest that interneuronal connectivity along with the cell-to-cell propagation of pathogenic α-syn is the major determinant for the pattern of Lewy body pathology [18,20,40,45].
In addition to brain homogenates and WT α-syn PFFs from both mice and humans, various modified forms of PFFs have been stereotaxically injected into the mouse brain. For example, phosphorylated S129 fibrils (p-PFFs) and phosphorylation-incompetent S129A fibrils were injected into the striatum to compare the spreading patterns and other features with those of WT α-syn PFF-injected mice. p-PFF-injected mice exhibited more α-syn inclusions in the substantia nigra pars compacta (SNpc) than WT α-Syn PFF-injected mice or S129A-PFF injected mice as early as 60 days postinjection, and these mice presented exacerbated pathology in the cortex [21]. Terada et al. [46] injected both C- and N-terminally truncated forms of human α-syn, including ΔC10 seeds, ΔC20 seeds, ΔC30 seeds, ΔN10 seeds and ΔN30 seeds, into the striatum. Mice injected with N-terminally truncated human α-syn PFFs, like mice injected with WT human α-syn PFFs, exhibited p-α-syn aggregates that were distributed throughout various brain regions, including the bilateral striatum, SN, amygdala, and anterior cingulated cortex, 3 months postinjection. Mice injected with C-terminally truncated PFFs displayed fewer p-α-syn inclusions than those injected with N-terminally truncated PFFs and exhibited inclusions only in the ipsilateral striatum, SN, and amygdala [46]. These findings indicate that the different truncated forms of α-syn also possess pathology-spreading capacities and phenotypic diversity.
Along with mice, rats have also been used to observe the propagation of endogenous α-syn upon the injection of PFFs. Intrastriatally injected adult Sprague-Dawley rats demonstrated that injections of both nonfibrillized recombinant α-syn and α-syn PFFs lead to significantly increased accumulation of p-α-syn inclusions, which are mainly found in the striatum, frontal and insular cortices, amygdala, and SNpc, 180 days postinjection [47,48], The process by which nonfibrillized α-syn induces aggregate propagation is not understood. The injection of PFFs into the SNpc in rats resulted in similar spreading patterns and dopaminergic neurodegeneration as injections into the dorsal striatum, showing a correlation between nigrostriatal inclusion spread and dopaminergic degeneration [35]. When Sprague-Dawley rats were injected with an AAV vector expressing human α-syn and then injected again with human α-syn PFFs into the SN 4 weeks later [45], florid p-α-syn pathological changes along with many features of PD, such as dopaminergic degeneration, hypertrophic microglia, T cell infiltration, and behavioral deficits, were noticed as early as 10 days after the injection of PFFs [45]. This indicates that the capability of PFFs to trigger α-syn pathology is enhanced by higher α-syn expression.
Interestingly, bilateral intracerebral injections of soluble Δ71–82 α-syn, a variant that cannot form fibrils, or fibrillar 21-140 α-syn in hippocampi of M20 mice also results in p-α-syn inclusions in many brain regions, such as the entorhinal cortex and other cortical regions, striatum, midbrain, and brainstem 2 and 4 months postinjection [49]. This raises the question as to whether fibril-forming ability is essential for triggering aggregate propagation.
PFF injection into the striatum of nonhuman primates resulted in the accumulation of p-α-syn in various brain regions and dopaminergic cell loss, along with microglial reactions [50]. An earlier study showed that the injection of Lewy bodies (LB)-enriched tissue extracts into the striatum of nonhuman primates also causes p-α-syn accumulation and dopaminergic cell loss in the SNpc, as well as dopaminergic terminal loss in the striatum [51].
Many studies on PFFs have shown neuronal loss along with inclusion spreading. However, the role of aggregate propagation in neurodegeneration remains elusive. In a study in which individual neurons were imaged for months after PFF injection in live animals, Osterberg et al. [39] showed that inclusion-bearing neurons selectively degenerate. This is one example of how PFF models can be used to address the relationship between aggregate propagation and neurodegeneration, and future studies will enhance our understanding of this subject.
SYNUCLEINOPATHIES IN PERIPHERAL PFF INJECTION MODELS
Based largely on Braak’s observations [52], there have been several attempts to generate models of propagation in mice by injecting PFFs into the olfactory bulb and peripheral regions. The injection of PFFs into the olfactory bulb of WT mice caused the conversion of endogenous α-syn into pathological aggregates, and the spreading of the inclusions is detected in more than 40 brain regions and subregions within 12 months postinjection [53]. These mice also exhibited neuronal loss in the anterior olfactory nucleus and deficits in olfaction [29,53]. The intramuscular (IM) injection of α-syn PFFs induced a rapid and predictable pathology in the CNS of M83 mice 2 to 3 months after injection [27,42]. The introduction of PFFs via the biceps femoris generated pathology similar to that seen in untreated aged M83 mice [27]. α-syn inclusion pathology is found in the spinal cord, brain stem, and midbrain structures but is relatively sparse in the cortex and in the sciatic nerve or muscle injection site [27]. IM injection resulted in spinal motor neuron degeneration and the loss of neuromuscular junctions [27], which suggests that PFFs can act through retrograde transport from the injection site [27,54]. In fact, the direct injection of PFFs into the sciatic nerve induced the spreading of the α-syn pathology throughout anatomically connected regions via axonal transport [54].
The injection of α-syn PFFs in the enteric neurons of the descending colon produced pathology in the brainstem of rats, but the progression ceased after 1 month postinjection [55]. In another study using the mouse gastric wall as the injection site, Uemura et al. [28] generated a model that exhibited α-syn pathology resembling that observed in the very early stage of PD. Injection into the gastric wall gave rise to p-α-syn-positive LB-like aggregates in the dorsal motor nucleus of vagus (dmX) after only 45 days. Retrograde propagation from the injection site to the dmX was abolished after vagotomy was performed, which demonstrates that the vagus nerve is the propagation route. No further rostrocaudal propagation beyond the dmX was observed up to 12 months postinjection, and no cell-type specificity of the affected neurons was reported [28]. In nonhuman primates, the injection of PFFs into the gastric walls and colon produced p-α-syn in both organs [55]. However, this study failed to detect the spreading of α-syn aggregates into the CNS.
In addition, the intraperitoneal introduction of α-syn PFFs efficiently generated α-syn pathology in the CNS of 4 out of 5 mice after 229 ± 17 days, while introduction via the intraglossal route generated CNS pathology, forming sarkosyl-insoluble aggregates and p-α-syn in both the brain and the spinal cord, in only 1 out of 5 mice after 285 days [30].
The extent of spreading of LB-like pathology into the CNS as a result of peripheral injection varies widely. This may depend on many factors, including but not limited to the histological structures of the injection sites, the size of the fibrils, and the amount of fibrils used, which depends on the injected organs. Considering the nonuniformity of the experimental conditions of different studies of peripheral injections, it is not justified to consider one injection site superior to another until more studies are carried out.
The search for the neuroanatomical pathways from the periphery to the brain through which α-syn pathology progresses is far from complete, and studies have shown that the progression of α-syn pathology does not always follow neuroanatomical pathways [27,56], although there have been studies showing that it does [42,54]. It is noteworthy that these studies were conducted under different conditions, and only fragmented parts of the neuroanatomical tracts were investigated, which might have yielded inconsistent results. The nature of endogenous α-syn, such as its protein expression level and its tendency to aggregate, is an important factor that might affect the experimental results. Higher endogenous levels of α-syn in Tg animals can accelerate the peripheral induction of pathology [27,49,54]. Additionally, the fact that the A53T mutant of α-syn is more prone to aggregation into amyloid in vitro might explain why A53T carriers show earlier and faster neuropathology and motor symptoms than their M20 peers that express WT α-syn [49,54].
PFF STRAINS
Although PFFs have common structural features that categorize them as amyloid fibrils, there are conformational variations. These conformational variants are referred to as strains because their biological effects are different. In vitro, strains have been generated under different conditions, and when they are injected into mice, the mice manifest distinct patterns of inclusions and pathological severity [57]. For example, pathological outcomes were investigated after different conformational variants of human α-syn aggregates, such as ribbons and fibrils, were introduced by intracerebral injection into the rat SNpc and by systemic injection [57]. The ribbons and the fibrils were generated under different salt concentrations [58]. Lewy bodies and Lewy neurite-like inclusions were present in dopaminergic neurons 4 months after the inoculation of the ribbons and fibrils; the inclusions were more abundant after the administration of ribbons. Although the ribbons induced more inclusions, more severe neurotoxicity was observed after fibril inoculation. The injection of ribbons led to the formation of glial cytoplasmic inclusions; however, the injection of fibrils did not, suggesting that differences in the strain can explain the differences in PD and MSA pathology. Aggregates were also found in cortical neurons and the spinal cord, with pronounced microglial responses after the intravenous injection [57].
Other strains were generated in the presence or absence of lipopolysaccharide (LPS). These strains showed distinct molecular features and degradation/internalization kinetics by neurons and microglia [59]. The injection of the LPS(-) PFFs into the striatum showed a strong and widespread pathology 6 months after injection, whereas the LPS(+) PFFs induced inclusions mostly in the striatum and the auditory cortex. These results suggest that exposure to environmental agents can lead to the generation of diverse strains with distinct properties, which in turn induce different synucleinopathies [59].
It has also been suggested that different α-syn strains have different abilities to crosstalk with tau pathology. The inoculation of two human α-syn PFF strains obtained either by de novo generation (one passage) or by repetitive seeding (2 to 4 passages) into the hippocampus of tau Tg PS19 mice resulted in distinct patterns of phospho-tau deposition in the brain [60].
NEUROIMMUNE RESPONSES IN PFF MODELS
Gliosis and neuroinflammation are other neuropathological features of PFF models. α-Syn aggregates derived from neurons interact with microglia [61] and astrocytes [62], trigger changes in gene expression and initiate glial inflammatory responses. Likewise, the injection of human or mouse α-syn PFFs into WT or Tg mice expressing human α-syn results in increased microglial activation and the release of proinflammatory mediators. Differences in the inflammatory profile depending on the site of injection, type of seed, and time point of pathology assessment have been reported [21,40,45,63].
In M83 mice, the inoculation of α-syn fibrils into the forebrain caused astrogliosis and microgliosis in the striatum, cortex and brainstem 90 days postinjection, as determined by glial fibrillary acidic protein (GFAP) immunostaining [20]. In a similar manner, massive astrogliosis and microgliosis occurred at the site of injection and the entorhinal cortex 4 months after the hippocampal injection of 21-140 α-syn fibrils in M20 Tg mice [64], a mouse model expressing human α-syn that does not develop synuclein pathology or neurological defects [49,65].
In a different study, Thakur et al. [45] used a combination of PFF injection and AAV-mediated overexpression of human α-Syn in dopaminergic neurons in the SN. This approach sped up the pathogenic process and produced a long-lasting inflammatory response. Microglial reactions were observed in animals exposed to α-syn PFFs alone and were augmented with adenoviral-mediated α-syn expression. Additionally, they observed a spatial correlation between activated microglia and neurons (and processes) containing p-α-syn. This model also showed CD8-positive and CD4-positive lymphocyte infiltration 3 weeks after injection.
The phosphorylation state of PFFs has been shown to have an impact on innate immune cell responses. Animals were injected with three types of fibrils, namely, WT PFFs, S129A-PFFs and phosphorylated PFFs, and cytometry analysis revealed differences in resident and infiltrated immune cells in the CNS. The injection of WT and S129A PFFs caused the infiltration of peripheral macrophages, while the injection of p-PFFs reduced the number of infiltrated immune cells. However, the latter injected induced a higher expression of NOS-expressing macrophages, which resemble a proinflammatory phenotype [21], suggesting that the structural characteristics of fibrils influence the activation of distinct immune cells. Cytokine expression changes have also been noted in PFF models. TNF-α levels were increased after PFF injection regardless of fibril type [21]. However, the levels of IL-10 were decreased after the injection of p-PFFs, suggesting different profiles in immune cell responses. In addition, the analysis of IFN-γ showed no changes for all PFFs [21].
Recently, another element of the innate immune system, the inflammasome, has been proposed to play a role in inflammation during the propagation of α-syn. Gordon and colleagues demonstrated that the NLRP3 inflammasome and the downstream elements cleaved caspase-1 and ASC are upregulated and activated in a PFF model mouse 30 days after injection and proposed the inhibition of NLRP3 as a therapeutic target for PD [66].
Inflammatory immune responses may be the cause rather than the consequence of neurodegeneration in PFF models. After intranigral injection in rats, the major histocompatibility complex (MHC) II response was induced as early as 24 hours postinjection, persisted over time, and was accompanied by CD163-positive cells, revealing the infiltration of scavenger macrophages from the periphery. In addition, a higher number of CD45High/Cb11b macrophages and CD4+ T cells appeared in the SN at 2 months, but there were no changes in the number of microglia, suggesting changes in microglial activation instead of an increase in proliferation. Because dopaminergic neuronal loss in the SN and a decrease in nigrostriatal projection was not apparent until 3 to 6 months postinjection, it seems that degeneration occurs due to the recruitment of peripheral immune cells expressing proinflammatory phenotypes triggered by MHC II activation [63].
Similarly, significant changes in MHC II expression in the SN have been shown to occur after intrastriatal injection, reaching a peak at 2 months after injection and decreasing during degeneration. Additionally, there are no differences in the number of microglia; however, morphological changes can be observed 3 months prior to neuronal loss. This suggests that inflammatory components, such as MHC II, are activated as a primary mechanism upon the presence of inclusions as an attempt to reduce aggregate burden; however, this might contribute to neuronal vulnerability and lead to degeneration [47]. A recent study also suggested that, in humans, immune responses are early events rather than late events in association with aggregate propagation. Olanow et al. [67] showed that microglial inflammatory responses precede the propagation of Lewy body pathology in human fetal tissues grafted into patient brains.
Signs of astrogliosis and microgliosis also appear when α-syn fibrils are injected through systemic routes. M83+/+ mice that underwent IM injection showed increased staining for Iba-1 and GFAP in inclusions residing in areas such as the pons and gray matter of the spinal cord [27]. Additionally, immunoreactivity of the microglial marker Cd11b was increased in the spinal cord 3 months after IM inoculation and progressively spread to the midbrain [42]. Additionally, RNA sequencing of spinal cord tissues from end-stage mice showed robust immune activation with alterations in microglia-specific genes such as Tgm1, Mpeg1, and Cs7 [42].
Given the findings on the infiltration of peripheral immune cells in the brain after the injection of α-syn [47,63], recent research aimed to assess the participation of peripheral inflammation in the progression of PD. For this purpose, a combination of intracerebral injections of PFFs and intravenous injections of LPS were used, and the results demonstrated higher microglia and leukocyte activation in the brains of mice treated with a combination than in those treated with fibrils alone. Similar results were obtained for monocytes in the brain and spinal cord, suggesting that the priming of monocytes can be a source of inflammatory cells after the systemic injection of fibrils, thus paving the way for the spreading of α-syn to the brain [68].
Moreover, the analysis of intraperitoneally and intraglossally injected M83+/-GFAP-luc revealed enhanced radiance in bioluminescence imaging in the brains of mice before the development of neurological abnormalities. Because the expression of luciferase is associated with the GFAP promoter, the results demonstrate astrocyte activation in the brain. Deposits of p-α-syn were also accompanied by an increase of Iba-1 and GFAP immunostaining [30].
Many PFF models exhibit progressive neuronal loss. In the intrastriatal PFF model [21], the progressive loss of dopamine neurons in the SNpc along with reduced dopamine levels 90 days postinjection were observed in injected WT mice [18,21,40,41,45]. Significant early neuronal loss in the anterior olfactory nucleus, but no mitral neuronal loss, was revealed after the injection of α-syn fibrils in the olfactory bulb [29]. Similar to mice, PFF-injected rats exhibited bilateral reduced striatal dopaminergic innervation, while dopamine was reduced only in the ipsilateral striatum 60 and 180 days postinjection [48]. In addition, the survival rate of injected Tg mice was significantly reduced compared to that of non-injected Tg mice, indicating the acceleration of symptom onset [20]. When human α-syn PFFs were injected into the striatum, striatal γH2AX and 53BP1 foci, indicators of DNA damage and the activation of the DNA damage response, were significantly increased 4 months postinjection [41]. According to Nouraei et al. [69], it is necessary to examine “regional differences in vulnerability to proteotoxic stress” to recognize differentially expressed molecules that either promote or obstruct endogenous defenses, and these further studies will help researchers overcome the challenges and limitations of using PFFs.
NEUROPHYSIOLOGY IN PFF INJECTION MODELS
Changes in synaptic function precede neurodegeneration in animal models overexpressing human α-syn [70]. These changes include impairments in neurotransmitter release [71-73], firing activity [74] and long-term potentiation [75], suggesting that pathogenic α-syn alters neuronal communications. Synaptic activity in PFF models has not yet been reported extensively. However, few studies have investigated the effects of exogenous α-syn on synaptic activity. Martin et al. [75] and Diógenes et al. [76] performed electrophysiological recordings after exposing mouse brain slices to α-syn oligomers and found impaired synaptic transmission and long-term potentiation in the hippocampus. These results suggest that extracellular α-syn can exert neurotoxic effects through a mechanism that affects synaptic activity. However, to date, evidence is scarce as to whether these ex vivo results represent what occurs in the brain during α-syn propagation through neural circuits.
In light of this, the work of Blumenstock and coworkers is noteworthy. They showed dendritic spine pathology in the neocortex using in vivo two-photon microscopy after the intracerebral injection of PFFs into the dorsal striatum [43]. p-α-Syn accumulated in the cortical areas starting in layers IV and V 30 days postinjection and spreading to all layers as propagation progressed. Spine loss and malformation in layer V was observed five months after PFF injection [43].
Neurotransmitter activity is also impaired, as shown by in vivo amperometry recordings after SN injection. Injected animals exhibited a reduction in release and reuptake rates in the striatum partly as a result of dopaminergic terminal loss. These changes were only observed when fibrils were combined with α-syn AAV overexpression [45].
Recently, electrophysiology recordings from slices from PFF-injected mice revealed dysfunction in synaptic transmission and plasticity. These animals exhibited long term potentiation (LTP) impairment in the striatal medium spiny neurons. Pharmacological intervention of currents showed that this phenomenon was caused by alterations in the molecular composition of N-Methyl-D-aspartic acid (NMDA) receptors containing Glu-N2A subunits [77].
How α-syn aggregates modulate synaptic activity before causing neuronal death still needs to be elucidated. Wu et al. [78], using primary hippocampal pyramidal neurons, demonstrated that PFF treatment reduces mEPSC, sEPSC and mIPSC in a time-and dose-dependent manner by affecting synapse formation; this was observed as a decrease in the colocalization of synapsin 1 and PSD-95 and a disruption of spine dynamics and morphology. It is worth mentioning that Wu and coworkers used a whole-cell patch clamp technique to investigate synaptic activity modulation after exposure to PFFs at the single-cell level [78]. Using this single neuron-based system, they reported decreased synaptic activity within 10 min following the direct administration of PFFs to the neuron and determined that synaptic dysfunction occurs in early stages of PD pathogenesis [78].
BEHAVIORAL CHANGES IN PFF INJECTION MODELS
Along with changes in neuropathology and neurophysiology, altered behavior has been observed in PFF models. In an intrastriatal injection model, PFF-injected mice showed decreased latency in the wire hang test 6 months postinjection, indicating a reduction in grip strength [18,66]. The injection of soluble α-syn monomers did not result in any significant deficits in the wire hang test [40]. In the rotor-rod test (conventionally referred to as the rotarod test), which assesses sensorimotor coordination and balance, PFF-injected mice showed impairments in motor function and balance 6 months postinjection [18,66]. Similarly, PFF-injected mice showed deficits in the balance beam test, which assesses motor function and balance [18]. However, Masuda-Suzukake et al. [40], used a similar procedure and observed similar pathological outcomes but was not able to replicate the behavioral phenotypes. The reason for this discrepancy is not clear [66].
The open field task is a widely performed sensorimotor test to analyze overall motor activity and locomotion, and it determines general activity levels, including exploration habits, which also represent emotional affect. PFF-injected mice did not show significant changes in overall motor activity during the open field test [18,40]. On the other hand, 8-month-old PFF-injected mice exhibited increased rotation behavior in the open field test [66], which indicates that there might be emotional abnormalities elicited by α-syn propagation [79,18]. When either exogenous monomeric or aggregated α-syn was injected into the mouse olfactory bulb, a shorter life span and neuronal transport to connected brain regions were observed [53]. Additionally, no pathological changes in α-syn and no motor- or anxiety-related deficits were observed, but significantly defective olfactory function was detected in several olfactory tests, including odor detection and odor retention tests [53]. Tg mice intramuscularly injected with PFFs presented α-syn inclusion pathology, astrogliosis, and microgliosis, as well as rapid, robust, and lethal motor phenotypes [27].
In contrast to the motor deficits and emotional abnormalities, learning and memory behavior was relatively spared in the PFF models up to 6 months after injection [18,40]. Whether this is due to the injection site (such as the striatum, olfactory bulb, and peritoneum) and/or the duration of pathological spreading remains to be determined.
CONCLUSIONS AND PERSPECTIVES
The spreading of aggregate pathology is thought to be mediated by cell-to-cell aggregate propagation, and the pathological and behavioral aspects of synucleinopathies have been recapitulated in PFF models. The pattern of aggregate spreading in PFF models depends on the type of seed used and the site of injection (Table 1, Figure 2). Regardless of these variables, PFF models induce progressive protein aggregation, neuronal loss, neuroinflammation, and behavioral phenotypes. These pathogenic changes stem from the progressive aggregation of endogenous α-syn proteins triggered by a single injection of a small amount of aggregates made of the homologous protein. Furthermore, PFF models are advantageous over pre-existing overexpression-based model of synucleinopathies due to the ability to control the spatiotemporal aspects of pathogenic events. Given these characteristics, many researchers consider PFF models to be the best available in vivo model systems for synucleinopathies and valuable tools for the study of disease pathogenesis as well as for therapeutic development. One of the weaknesses of PFF models is the variability of results, especially among different research groups. Established guidelines for PFF preparation and injection protocols would improve the robustness and reproducibility of the results. Notwithstanding, we foresee critical contributions of PFF models to advancements in understanding important questions in the field, such as the intricate interaction between protein aggregation, neuroinflammation, and neurodegeneration. Using these models to spatiotemporally control aggregate propagation, we can also address the impact of aggregate pathology on neural circuit dysfunction.

Injection methodologies from recent studies

Propagation maps for the different injection sites and preformed fibrils (PFFs). The dots represent phospho-α-syn (p-α-Syn) inclusions in different areas of the brain and spinal cord after PFF inoculation. The dots located under each panel indicate that the specific distribution of aggregates was not detailed for that area in the original article. WT: wild type, p.i: post injection.
Notes
Conflicts of Interest
S-JL is the founder and CEO of Neuramedy Co., Ltd. HKC, H-AH, and DP-A are not related to Neuramedy Co., Ltd.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIT, NRF-2018R1A2 A1A05078261 and NRF-2018R1A5A2025964). Dayana Pérez-Acuña was supported by the CONICYT PFCHA/DOCTORADO BECAS CHILE/ 2018-72190194.