Neurodegeneration with Brain Iron Accumulation: Diagnosis and Management
Article information
Abstract
Neurodegeneration with brain iron accumulation (NBIA) encompasses a group of inherited disorders that share the clinical features of an extrapyramidal movement disorder accompanied by varying degrees of intellectual disability and abnormal iron deposition in the basal ganglia. The genetic basis of ten forms of NBIA is now known. The clinical features of NBIA range from rapid global neurodevelopmental regression in infancy to mild parkinsonism with minimal cognitive impairment in adulthood, with wide variation seen between and within the specific NBIA sub-type. This review describes the clinical presentations, imaging findings, pathologic features, and treatment considerations for this heterogeneous group of disorders.
INTRODUCTION
Neurodegeneration with brain iron accumulation (NBIA) comprises a heterogeneous group of inherited neurodegenerative disorders collectively characterized by extrapyramidal movement disorders and abnormal iron accumulation in the deep basal ganglia nuclei of the brain. Ten NBIA genes have been identified to date: eight are autosomal recessive, one is autosomal dominant, and one is X-linked dominant (Table 1). Prevalence data is incomplete, but all forms of NBIA are considered to be “ultra-rare” with less than 1/1000000 affected. Four types of NBIA predominate as shown in Figure 1: pantothenate kinase-associated neurodegeneration (PKAN); phospholipase A2-associated neurodegeneration (PLAN); mitochondrial membrane protein-associated neurodegeneration (MPAN); and beta-propeller protein-associated neurodegeneration (BPAN). This distribution may shift slightly over time as new cases of more recently discovered mutations such as WDR45 grow in number.
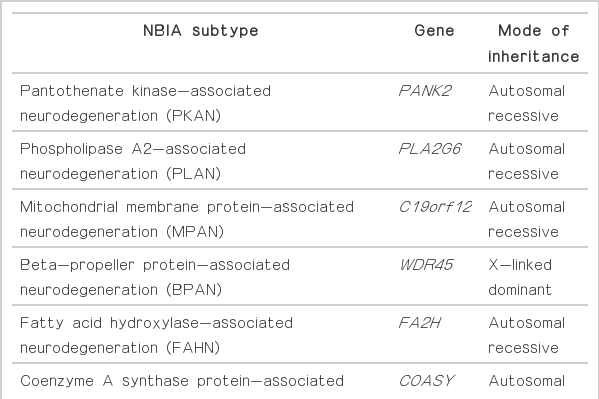
The ten forms of NBIA described to date, with name, acronym, mutated gene, and mode of inheritance
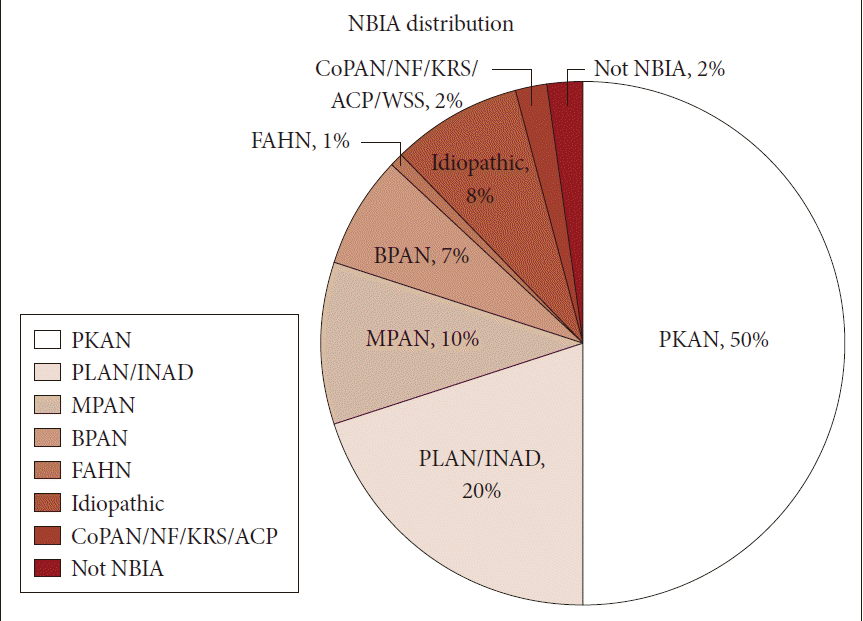
Distribution of NBIA subtypes in the North American database. NBIA: neurodegeneration with brain iron accumulation, PKAN: pantothenate kinase-associated neurodegeneration, PLAN: phospholipase A2-associated neurodegeneration, INAD: infantile neuroaxonal dystrophy, MPAN: mitochondrial membrane protein-associated neurodegeneration, BPAN: beta-propeller protein-associated neurodegeneration, FAHN: fatty acid hydroxylase-associated neurodegeneration, CoPAN: Coenzyme A synthase protein-associated neurodegeneration, NF: neuroferritinopathy, KRS: Kufor-Rakeb syndrome, ACP: aceruloplasminemia.
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION (PKAN)
Most of the cases reported in the historical literature as “Hallervorden-Spatz disease” were probably PKAN, but almost certainly included other forms of NBIA, an important observation when trying to glean insights about PKAN from the older literature. Julius Hallervoden and Hugo Spatz were German neuropathologists whose work derived from pathological samples obtained under the Nazi program of active euthanasia of individuals with physical and intellectual disabilities. The PANK2 gene discovery [1] provided not only an opportunity to establish a naming convention for NBIA, but also the catalyst to dishonor and abandon the eponym [2,3]. PKAN represents the most prevalent form of NBIA, accounting for half of the patients in our research registry (Figure 1). The PANK2 gene encodes pantothenate kinase 2, which phosphorylates vitamin B5, N-pantothenoyl cysteine, and pantetheine in the first key regulatory step of coenzyme A biosynthesis, and thus is essential for intermediary and fatty acid metabolism.
Pantothenate kinase-associated neurodegeneration has traditionally been divided into early-onset, rapidly progressive “classic” disease and a more slowly-progressive “atypical” form that has a later onset and slower progression [4], though there certainly are cases that fall somewhere between, combining features of both forms. In classic PKAN, the child presents before the age of six, usually with a change in gait and falls, sometimes superimposed on a history of mild developmental delay. The neurologic exam at presentation generally reveals evidence of dystonia, with the lower limbs predominantly affected: a striatal toe sign may appear intermittently during the exam, or there may be dystonic posturing of the child’s foot during gait testing. Corticospinal tract signs may also be present, with spasticity, brisk reflexes and extensor plantar responses seen. There may be a history of poor night vision, and formal ophthalmologic evaluation often reveals pigmentary retinopathy with an abnormal electroretinogram, bilateral Adie’s pupil, and various abnormalities of pursuit and saccadic eye movements [5]. Pathognomonic abnormalities are seen on MRI imaging of the brain, even early in disease (see Imaging, below). Acanthocytes may be seen on prepared blood smears [6].
The progression of classic PKAN is not linear, but rather stepwise, with periods of rapid decline occurring, usually without clear precipitant. As the trunk becomes involved, the child may exhibit a striking opisthotonic posturing that is a hallmark of classic PKAN. Limb dystonia may be curiously asymmetric despite the symmetry of the pallidal lesions on MRI (Figure 2) but impairment of gait and balance is universal and results in most children needing a wheelchair before adolescence. Speech and swallowing are affected as the disease involves the bulbar musculature, causing nutritional compromise and presenting a risk of aspiration pneumonia. Some children with classic PKAN succumb to complications of their disease in the first decade, but many survive into adulthood, albeit with severe disability.
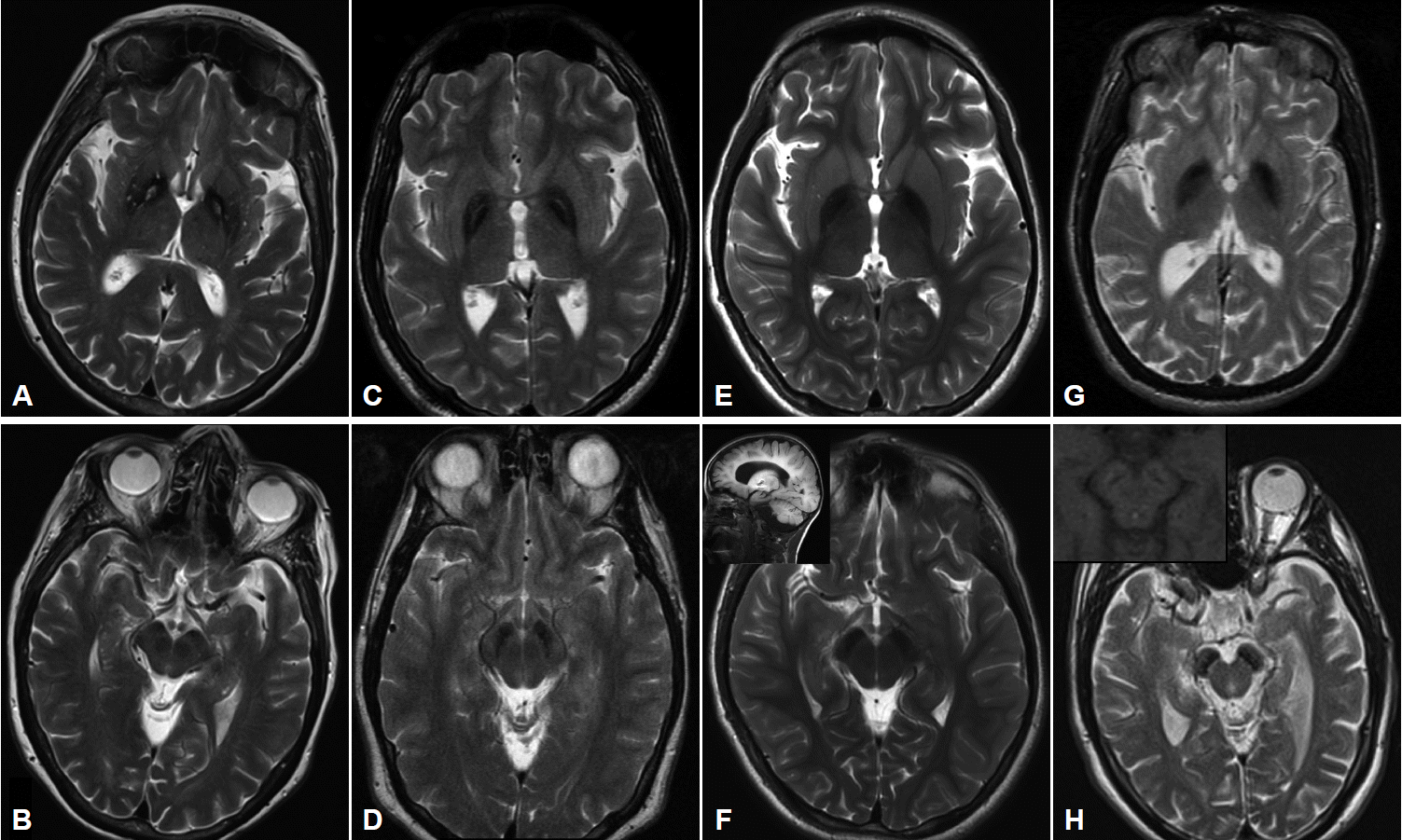
Imaging characteristics of the four major subtypes of NBIA. All images performed on 3.0T magnet except (G) and (H) which were performed on 1.5T. A: T2-weighted imaging in PKAN shows GP hypointensity indicating iron accumulation with an anteromedially-located area of hyperintensity, the so-called “eye of the tiger”. B: SN in same patient showing hypointensity in medial aspect of nucleus. C: T2-weighted sequence in a patient with MPAN showing pallidal hypointensity with hyperintense streaking in the region of the medial medullary lamina. Depending on the cut, this may be mistaken for an “eye of the tiger” characteristic of PKAN. D: The SN in the same patient also demonstrates iron accumulation. E: T2 sequence of GP and (F) SN in a 9 yo child with PLAN showing evidence of iron accumulation. Imaging performed earlier in the disease course had shown no signal changes. Inset in (F) showing cerebellar atrophy in the same child. G: T2 imaging showing the GP in a young adult with BPAN, after the onset of parkinsonian symptoms. In (H) note the marked hypointensity in the SN, and, in the inset in (H), the same region on T1 weighted sequence showing the characteristic hyperintense “halo” thought to represent neuromelanin release from degenerating neurons. PKAN: pantothenate kinase associated neurodegeneration, MPAN: mitochondrial membrane protein-associated neurodegeneration, BPAN: beta-propeller protein-associated neurodegeneration, PLAN: phospholipase A2-associated neurodegeneration, GP: globus pallidus, SN: substantia nigra, NBIA: neurodegeneration with brain iron accumulation.
When PKAN presents later in life, signs and symptoms are far more heterogeneous. The teenager or young adult with atypical PKAN may present with a change in speech patterns, manifesting as stuttering, a Parkinsonian-type palilalia or hypophonia, spasmodic dysphonia, or dysarthria due to oropharyngeal dystonia. A new tic disorder may appear, or there may be a marked worsening of tics attributed earlier in childhood to Tourette syndrome. Neuropsychiatric symptoms are also common in atypical PKAN. Mood lability, impulsivity, non-specific behavioral changes, and obsessive-compulsive features may be early signs and may be attributed to typical changes of adolescence until other neurologic signs appear, prompting further workup. Typical motor signs of illness may not be apparent until later in disease, with most patients exhibiting mixed dystonia and parkinsonism and varying degrees of spasticity. The extrapyramidal motor symptoms in atypical PKAN show an age dependency, with adolescents demonstrating more dystonia than parkinsonism, while the opposite pattern is observed in patients presenting in their 20s, who show bradykinesia, rigidity, freezing of gait and postural instability on exam; a rest tremor is less commonly a feature. Action-induced dystonias are frequently seen in atypical PKAN and may offer an important clue to diagnosis. Oromandibular dystonia triggered by eating and speaking is particularly unusual among the secondary dystonias, chorea-acanthocytosis, the related McLeod acanthocytosis syndrome, and neuroferritinopathy being the only other disorders where eating dystonia is a prominent feature. Writer’s cramp and action-induced dystonic tremors may also be observed. Pigmentary retinopathy is rare in atypical PKAN, though extra-ocular movement abnormalities are common.
The progression of atypical PKAN is much slower than classic disease, and many individuals have a normal lifespan. Our own observation is that the rate of decline tends to be steeper following the onset of symptoms, but then tends to stabilize and change only minimally over years.
Cognition may be abnormal in PKAN, but intellectual disability is not a universal part of the disease as suggested by the early “Hallervorden-Spatz” literature and cognitive decline over time is not typical. The younger the onset of disease, the greater the degree of cognitive impairment [7]; this may be because the disease process in very early-onset PKAN disrupts normal brain development. The severity of symptoms may also impact performance on standardized cognitive tests. This, in turn, may lead to overestimation of intellectual disability; a study of children with PKAN undergoing deep brain stimulation surgery found post-surgery improvements in cognitive test performance concomitant with improvement in dystonia symptoms [8].
Imaging findings in PKAN
MRI imaging in PKAN is distinctive enough to be diagnostic in most cases. T2 weighted sequences through the basal ganglia reveal the globus pallidus to be hypointense with an anteromedially-placed region of hyperintensity, the so-called “eye of the tiger” sign (Figure 2). The substantia nigra shows normal to mildly hypointense signal. Although these findings in an individual with characteristic signs on exam are highly predictive of the presence of PANK2 mutations, the finding is not absolutely sensitive or specific: rare mutation-positive cases are described in which there is no “eye of the tiger” on MRI [9,10]. Conversely, “eye of the tiger” mimics may be seen in MPAN, carbon monoxide poisoning survivors, multiple system atrophy, neuroferritinopathy, and other unknown diagnoses [11,12].
Pathology of PKAN
The pathology of PKAN is much more circumscribed than previously thought, being largely limited to the globus pallidus, in contrast to other forms of NBIA [13]. In gross section, the presence of iron is evidenced by a frankly rusty discoloration of the globus pallidus but not other structures; this is confirmed microscopically with iron-specific stains that reveal the iron to have a perivascular distribution. A central area of neuronal depletion and tissue rarefaction corresponds to the “eye of the tiger” seen on MRI. Two populations of ubiquitin-positive spheroids are seen. Larger pleiomorphic structures thought to represent degenerating “ghost” neurons are concentrated in the globus pallidus but are also noted in the putamen and internal capsule. Smaller eosinophil structures represent the classic neuroaxonal spheroid seen in various forms of NBIA; these are sparsely located in the globus pallidus, corpus callosum and subcortical white matter and stain for amyloid precursor protein as well as ubiquitin.
Treatment considerations in PKAN
Currently, treatment is symptomatic. Dystonia and spasticity are usually managed with anticholinergics, benzodiazepines and other anti-spasticity agents such as baclofen, which may be delivered intrathecally. Botulinum toxin injections can also provide targeted relief of dystonia and spasticity. Deep brain stimulation has shown promise, but studies are limited to individual case reports, small case series [14] and a retrospective study [15] which included non-PKAN cases, challenging the generalizability of the results. The ongoing involvement of physical, occupational and speech therapists can delay complications of disease.
One of the most challenging problems for the patient, family and clinician in PKAN is dystonic crisis or “dystonic storm”. This severe exacerbation of dystonia is seen mostly in children with classic disease and can be life-threatening. It can occur without an obvious precipitant, but the child should be screened for infection, fecal impaction, and occult fractures to be certain there is not a treatable cause. The torsional stress created by the severe dystonia of classic PKAN can result in occult fractures of long bones, especially in children who are no longer weight-bearing and may be osteopenic. The treatment of dystonic storm is very challenging; no controlled studies have been published but treatment strategies have been reviewed in two publications [16,17].
The contribution of iron accumulation to the core pathophysiology of the disease remains in question [18], but until that question is answered, iron chelation remains under investigation as a disease-modifying approach. A preliminary study of deferiprone, an iron chelator that readily crosses the blood-brain barrier, showed robust reduction of brain iron on brain MRI in PKAN patients, but no measurable benefit in clinical disease outcomes [19]. A double-blind, placebo-controlled international multicenter clinical trial is underway to more rigorously investigate the drug’s benefit (https://clinicaltrials.gov/ct2/show/NCT01741532?term=deferiprone+PKAN&rank=1). A rational therapeutic approach aimed at bypassing the enzymatic defect with a novel compound is also under investigation.
PHOSPHOLIPASE A2-ASSOCIATED NEURODEGENERATION (PLAN)
Mutations in the calcium-independent phospholipase A2 gene PLA2G6, thought to play a critical role in cell membrane phospholipid homeostasis, are responsible for PLAN. When PLAN presents in early childhood, it is called infantile neuroaxonal dystrophy (INAD), a term that originates from the hallmark pathologic finding of dystrophic axons found on nerve or conjunctival biopsy; before genetic testing was available, this finding confirmed the diagnosis. While we have tried to rationalize the rules surrounding nomenclature in the NBIA field by naming each disease using the “(mutant protein)-associated neurodegeneration” convention, INAD is firmly established in the literature and continues to be routinely used for the infantile-onset form of PLAN.
Classic INAD is a devastating syndrome of neurodevelopmental regression. It presents between 6 months and 3 years of age, initially with slowing or cessation of development, followed by progressive loss of previously acquired milestones in all domains. Truncal hypotonia appears early in the course, sometimes leading to consideration of spinal muscular atrophy in the differential diagnosis. Optic atrophy, strabismus and nystagmus are prominent and are accompanied by visual impairment leading to eventual blindness in many cases. Hypotonia and areflexia is replaced by spastic tetraplegia as the disease advances, a result of early peripheral denervation from an axonal sensorimotor neuropathy and later pyramidal dysfunction. Electroencephalography is abnormal with frontal-predominant fast rhythms and sometimes overt epileptiform discharges. Though seizures have been described as rare and only occurring late [20] a recent review of 25 children with INAD noted epilepsy in 17% [21]. Most children with classic INAD die before the age of 10.
When the onset of PLAN is later in childhood, and for a minority of patients initially presenting as classic INAD, the disease’s presentation is more variable and its progression slower. These children are said to have “atypical infantile neuroaxonal dystrophy” (aNAD). Their picture differs from the classic form of the disease: cerebellar dysfunction with gait ataxia and dysarthria are more evident, and hypotonia and areflexia tend to predominate, rather than spasticity. Additionally, dystonia may be present. The cerebellar presentation in aNAD may simply be a function of timing of the disease in relation to development: children with classic INAD lose their ability to walk and talk so early in the disease course that signs of cerebellar disease may never be clinically apparent. Cognition is impaired: the child may be initially assessed as having a static encephalopathy, but progressive cognitive decline eventually becomes apparent, sometimes the first clue that the child or teen has a neurodegenerative disorder.
The PLA2G6 gene discovery and advances in molecular genetic technology led to the recognition of an adult form of PLAN presenting with dystonia-parkinsonism and spasticity, along with cognitive and psychiatric features [22]. Cerebellar features, denervation on electromyography, and fast rhythms on electroencephalography are less common in adult-onset patients than in INAD or aNAD. The parkinsonism is often nicely responsive to dopaminergic medications, but motor fluctuations tend to emerge quickly and complicate management.
Imaging findings in PLAN
Cerebellar atrophy is the most common neuroimaging finding in both INAD and aNAD. It is present in the majority of patients even when imaged early in disease, and in virtually 100% of well-established INAD cases. It is an important clue to the diagnosis in a child undergoing workup for psychomotor regression. Additional features seen on MRI include diffuse T2 white matter hyperintensities, and thinning of the corpus callosum and optic chiasma. The imaging of late-onset PLAN is more variable in its features: cerebellar atrophy is inconsistently present though cerebral volume loss may be evident, especially later in the disease. In all forms of PLAN, iron accumulation, manifesting as T2 hypointensity, may be absent or subtle early in the disease course and may never become evident in INAD and aNAD; susceptibility weighted imaging (SWI) may reveal iron not apparent on standard T2 sequences. Iron is generally evident in the globus pallidus on MRI imaging in late-onset patients presenting with clear-cut motor signs of dystonia and parkinsonism (Figure 2).
Pathology of PLAN
Pathologic studies are limited to case reports and small case series. Consistent with the MRI findings during life, cerebellar and cortical atrophy may be observed on gross pathology, with widespread neuronal loss and gliosis evident microscopically. Dystrophic axonal spheroids, historically the hallmark diagnostic finding, are seen as eosinophilc swellings in peripheral nerves, spinal cord, brainstem and basal ganglia. A more recently recognized pathologic feature of PLAN is the prominent Lewy body pathology involving the basal ganglia and neocortex, particularly striking in disease of later onset and longer duration. Tau pathology may also be observed with hyperphosphorylated neurofibrillary tangles and neuropil threads [23,24].
Treatment considerations in PLAN
Like other forms of NBIA, treatment of PLAN is geared towards relief of symptoms and prevention of complications. Standard medications to treat seizures, spasticity, dystonia, and parkinsonism are employed, though treatment with levodopa of adult-onset dystonia-parkinsonism is complicated by early motor fluctuations and exacerbation of neuropsychiatric symptoms. Physiotherapy early in INAD/aNAD may delay or prevent contractures. Gastrostomy tube placement supports nutritional status as the disease advances.
Prospects for treatment are perhaps more immediate in PLAN than in some other forms of NBIA, in part because mouse models recapitulate important aspects of the disease, providing a mechanism to test rational therapeutics. Although no clinical trials are yet underway, mouse studies have revealed reduced incorporation of docosahexanoic acid into the mutant mouse brain using novel imaging techniques [25–27], as well as derangements in calcium signaling [28], suggesting future directions for both imaging biomarker development and therapeutics. Perhaps most encouraging, a “proof of concept” study testing a gene therapy approach is underway in the mouse model [29].
MITOCHONDRIAL MEMBRANE PROTEIN-ASSOCIATED NEURODEGENERATION (MPAN)
Mitochondrial membrane protein-associated neurodegeneration is caused by mutations in c19orf12. The protein’s function is poorly understood, but preliminary functional experiments and its sub-cellular association with the mitochondrial membrane suggest a role in cellular energetics and fatty acid metabolism.
Clinical findings in MPAN
Mitochondrial membrane protein-associated neurodegeneration typically presents in the first decade of life, but may also have its onset in early adulthood. In childhood, development of a spastic gait with extensor plantar responses is typically the earliest sign, commonly accompanied by optic atrophy, learning difficulties, dysarthria, and sometimes behavioral and psychiatric features. Dystonia, when present, tends to be limited to the feet and hands. The initial presentation in adulthood is more variable, but typically manifests with cognitive and behavioral changes, parkinsonism and mixed gait disorders [30,31].
Generally, the disease progresses slowly, and most individuals with childhood onset survive into their 20s or beyond. However, some individuals may exhibit rapid and terminal progression, either immediately following the onset of disease or after a period of slow decline. Interestingly, the literature and our own experience suggest that this occurs primarily in adult-onset patients [31,32]. This is in contrast to PKAN and PLAN, where rapid progression is associated with childhood-onset disease.
As the disease progresses, lower motor neuron signs may emerge, particularly in childhood-onset patients. Clinically, this manifests as loss of deep tendon reflexes, muscle weakness and sometimes atrophy; electrophysiologically, as a motor neuronopathy/axonopathy [31,33–35]. The combination of upper and lower motor neuron findings may raise the question of amyotrophic lateral sclerosis [35,36]. Cognitive decline appears to be universal in MPAN, sometimes accompanied by disabling psychiatric features; most patients are overtly demented by mid-stage disease. Bowel and bladder incontinence are common; it is not clear whether this is due to an autonomic neuronopathy or simply a non-specific feature associated with progressive dementia. Dysphagia is also common later in disease; this may lead to aspiration pneumonia.
Imaging findings in MPAN
Pallidal and nigral iron accumulation is evident on brain MRI on T2 and GRE sequences, variably accompanied in some patients by hyperintense streaking of the globus pallidus in the region of the medial medullary lamina (Figure 2). This may be interpreted as an “eye of the tiger” leading to an erroneous radiologic diagnosis of PKAN. Cortical and cerebellar atrophy may be seen in more advanced disease, and T1 hyperintensity in the caudate and putamen has been reported [31,35,37].
Pathology of MPAN
Pathologically, MPAN is a synucleinopathy, exhibiting a remarkable burden of Lewy bodies and Lewy neurites not only in the basal ganglia but also in the neocortex [31]. Cortical Lewy body pathology in MPAN exceeds that seen in sporadic Parkinson disease by 40-fold. Even Lewy body dementia, the archetypal cortical Lewy body disease, does not rival MPAN in this regard. Axonal spheroids, thought to represent dying neurons, are seen both peripherally and centrally. Iron deposits are seen in the globus pallidus and to a lesser extent in the substantia nigra; little or no iron is seen in the cortex. Neuronal loss and gliosis are especially prominent in the substantia nigra.
Treatment considerations in MPAN
Like all NBIAs, treatment is symptomatic. Anti-spasticity agents such as baclofen may be helpful and allow patients to maintain ambulation for a longer period. Trihexyphenidyl may provide relief when dystonia is problematic. Even when parkinsonism dominates the motor dysfunction, dopaminergic compounds seem to offer only modest relief at best, and may precipitate adverse neuropsychiatric effects; dopamine agonists in particular should be used with caution. There are no reports of deep brain stimulation in MPAN, but the prominent cognitive decline in the disease would strongly argue against its use. Treatment of bladder incontinence ideally should be guided by urodynamic studies and may need to be reassessed as the disease progresses, particularly if patients develop prominent lower motor neuron signs when urinary retention and overflow incontinence may replace a hyperactive detrusor. Standard bowel regimens may lessen the risks of impaction and fecal incontinence. A gastrostomy tube may maintain nutritional status and prolong life as dysphagia progresses.
When present, psychiatric features such as emotional lability, agitation, hallucinations, and compulsive behaviors can be particularly challenging for families. The close involvement of a psychiatrist familiar with treating cognitively-impaired patients is ideal. For patients with neuropsychiatric complications and prominent parkinsonism in particular, the choice of antipsychotic agent must be made with care and dosed cautiously.
BETA-PROPELLER PROTEIN-ASSOCIATED NEURODEGENERATION (BPAN)
Beta-propeller protein-associated neurodegeneration is unique among the NBIAs in its mode of inheritance, its presumed pathophysiology, its biphasic clinical profile, and its distinctive imaging characteristics. The only X-linked form of NBIA to date and a rare example of X-linked dominant inheritance, BPAN appears to have two distinct phases to its course, comprising a syndrome of global development delay with seizures, pyramidal signs and disordered sleep in childhood, followed by a decline in adulthood with the development of parkinsonism, dystonia and dementia [38]. Prior to the discovery of the causative gene, BPAN was described as “static encephalopathy with neurodegeneration in childhood” (SENDA), but it has now been named according to the established naming convention. The disease is caused by mutations in WDR45, which encode for a beta-propeller protein with a presumed critical function in autophagy based on the yeast homolog and preliminary functional work in humans [39,40]. Although dysregulation of autophagy has been proposed as having a role in neurodegeneration, BPAN is the first disorder in which a direct connection has been made.
The X-linked dominant pattern of inheritance means that most patients with BPAN will be female. The majority of BPAN cases arise as a result of de novo mutations early in development. In theory, a female with BPAN could arise as the result of either germline or somatic mutations; however, the severity of the phenotype means that most girls with BPAN will never reproduce. Skewing of X-inactivation could account for a mild phenotypes in females, though, and such cases are beginning to be reported [41]. Because affected males have only one copy of the X chromosome, the disease would be predicted to be male-lethal unless the mutation occurs post-zygotically; however, a recent report describes germline mutations in monozygotic twins and their older brother, all of whom exhibited intellectual disability and seizures [42]. The boys’ mother harbored the same mutation but was reported to be phenotypically normal, suggesting that perhaps the transmitted mutation had only minimal effects on function, allowing male survival despite hemizygosity. Clearly, the BPAN story is still unfolding with more surprises to come.
Clinical features of BPAN
The child with BPAN may initially appear normal, but it quickly becomes apparent that developmental milestones are not being attained on schedule. Motor milestones are delayed: the child may exhibit toe walking or a broad-based, ataxic gait. Intellectual disability is marked: little expressive language is acquired, with most children attaining only a few spoken words. Seizures are common, and may represent a major source of morbidity for the child with BPAN. On one end of the spectrum is the child who has only one or two seizures during childhood, none at all, or only febrile seizures; these children may require no treatment with anti-epileptic drugs. Other children, however, exhibit a picture of a severe syndromic epilepsy, with multiple seizure types present (partial complex, generalized tonic-clonic, atonic and/or myoclonic) that may be refractory to therapy. Some children exhibit sleep disorders, including hypersomnolence, hyposomnolence, shortened sleep latency, and abnormal rapid eye movement sleep. The presence of hand stereotypies in some children with BPAN, along with marked abnormalities in language development, disordered sleep, and seizures may have earned them a diagnosis of “atypical Rett syndrome” before mutations in WDR45 are found.
Children with BPAN make slow developmental gains in childhood, though remaining far behind their developmentally-normal peers. During the teenage years or early adulthood, signs of parkinsonism emerge, sometimes fairly precipitously. The posture becomes stooped, movements slow, and the gait changes, with shortened step length and freezing. While these symptoms are responsive to dopaminergic medications, sometimes robustly so, the benefit tends to be short-lived, with brittle levodopa-induced dyskinesias or dystonia quickly complicating management. In addition to the motor signs, a progressive decline in cognitive function becomes evident and previously-learned skills are lost. Challenging new behaviors may appear, often exacerbated by dopaminergic drugs. Though the decline may be gradual, eventually all patients lose ambulatory ability and become profoundly demented.
Imaging of BPAN
Imaging in early childhood is typically unremarkable. Later in the course, particularly as parkinsonism becomes evident on exam and prompts further diagnostic studies, a characteristic pattern appears on MR imaging. On T2 sequences, the globus pallidus, substantia nigra and cerebral peduncles become hypointense consistent with iron accumulation in these structures; these findings are more apparent and seen earlier in the disease with the use of iron-sensitive sequences such as T2* and susceptibility-weighted imaging. The hypointensity is most pronounced in the substantia nigra where it appears as a discrete linear streak. This same area on T1 sequences is surrounded by a hyperintense “halo” extending to the cerebral peduncles, thought to represent neuromelanin release from degenerating neurons (Figure 2) [38,43,44]. Derangements in white matter architecture and impaired pallidal and nigral metabolism have been demonstrated in a single adult BPAN case using diffusion tensor imaging and MR spectroscopy [44]. Other findings that may be seen on MRI include thinning of the corpus callosum, cerebellar atrophy and more global atrophy as the disease advances [38,40].
Pathology of BPAN
Grossly, cerebellar atrophy and thinning of the cerebral penduncles is apparent [38]. The iron accumulation can be seen in the unstained brain as a rusty discoloration in the substantia nigra. The iron is much less prominent in the unstained globus pallidus, but both structures show strong reaction when treated with iron-specific stains such as Prussian blue. Despite the prominent parkinsonism seen clinically, BPAN does not demonstrate the α-synuclein pathology seen in idiopathic Parkinson disease; rather, tau positive neurofibrillary tangles are seen in the cortex, putamen, hippocampus and hypothalamus. Axonal spheroids, a pathologic feature representing dying neurons and common to many forms of NBIA, are readily seen in the substantia nigra and globus pallidus, as well as in the medulla, pons and thalamus.
Treatment considerations in BPAN
As the most recent form of NBIA to be characterized, we are only just starting to understand the phenotypic spectrum of BPAN and define optimal approaches to management. In childhood, the most challenging problem is refractory seizures. Although only present in a minority, these BPAN patients should have the close involvement of a pediatric specialist experienced in the management of severe syndromic epilepsies. In adulthood, the parkinsonism can be treated successfully with dopaminergic medications, although as mentioned, motor fluctuations and dyskinesias pose problems and the drug benefit is not durable. Dopamine agonists might be predicted to have adverse neuropsychiatric effects in BPAN where cognitive impairment is a prominent part of the phenotype [45]; however, we have not seen that in our own clinic.
Preliminary work to create a mouse model of BPAN is yielding promising results [46]. As we begin to understand the role of autophagy in the disorder, we anticipate the development of rational therapeutics targeting the underlying defect.
Our own observations of the phenotype in BPAN to date are based on a highly homogeneous cohort selected specifically for gene discovery efforts. As more patients are found earlier in childhood through whole exome sequencing, we expect to see greater phenotypic variability, particularly since timing of somatic mutation and X-inactivation patterns likely influence the phenotype.
OTHER FORMS OF NBIA
Fatty acid hydroxylase-associated neurodegeneration (FAHN)
The FA2H gene product is responsible for hydroxylating fatty acids and plays a key role in myelin production in the central nervous system and possibly in cell cycle regulation [47]. Mutations in the gene were originally described as causing leukodystrophy and a complicated form of hereditary spastic paraplegia (HSP35) [48–50]. Subsequent investigations revealed the presence of pallidal T2 hypointensities in some patients harboring FA2H mutations and led to fatty acid hydroxylase-associated neurodegeneration’s (FAHN’s) designation as a subtype of NBIA [51]. The disease usually presents in the first decade of life with gait difficulties and falling. Progressive spasticity, dystonia and cerebellar dysfunction contribute to disability, leading to loss of ambulation in many cases, along with dysarthria and dysphagia. Optic atrophy is frequently present on exam, with variable degrees of accompanying visual impairment. Most children exhibit progressive cognitive decline; seizures are less consistently a feature. An axonal neuropathy has been described in one family [52]; further study is needed to see if this is a consistent part of the phenotype. In addition to evidence of iron accumulation in the globus pallidus (which is not always present), MR imaging of the brain reveals T2-bright white matter lesions, thinning of the corpus callosum, and progressive atrophy of cerebellum, pons, medulla and cord [53].
Many of FAHN’s clinical features mirror those of INAD/aNAD; thus, a child suspected of having PLAN but without mutations in PLA2G6 should be tested for FAHN. Like other forms of NBIA, the phenotypic spectrum of the disease is expected to broaden as more patients are discovered through whole exome sequencing.
COASY protein-associated neurodegeneration (CoPAN)
COASY protein-associated neurodegeneration (CoPAN) joins PKAN as the second inborn error of coenzyme A metabolism. CoPAN manifests in the first decade of life with gait difficulties and mild cognitive impairment. Oromandibular dystonia, dysarthria, and progressive spasticity follow, along with the appearance of an axonal neuropathy. The emergence of parkinsonism further adds to the disability. MRI demonstrates non-homogenous T2 pallidal hypointensity with a region of medial hyperintensity that is reminiscent of the “eye of the tiger” sign seen in PKAN. Although only two families with CoPAN have been reported thus far [54], the discovery that mutations in the gene encoding Coenzyme A synthase (COASY) could cause a form of NBIA confirms the importance of the CoA pathway in neuronal health.
Neuroferritinopathy
Mutations in the gene encoding ferritin light chain protein cause neuroferritinopathy, a dominantly-inherited syndrome of chorea, dystonia, parkinsonism, cognitive decline and low serum ferritin that typically presents in mid-life. A majority of the cases described in the literature are clustered geographically in the Cumbrian region of Britain and have a common 460insA mutation [55], suggesting a founder effect. Single families with private mutations and slightly different phenotypes have been described in Europe, North America, and Japan. Neuroferritinopathy can be distinguished from Huntington disease by its prominent action-induced orofacial dystonia, asymmetric presentation, late cognitive decline and by the presence of abnormal iron accumulation on brain MRI in the caudate, putamen, thalamus, globus pallidus, substantia nigra and red nucleus. Cystic change of the caudate and putamen occurs late in the disease [56,57]. Neuroferritinopathy differs from other forms of NBIA by its dominant inheritance pattern, uncommon presentation in childhood, and distinctive pattern of iron accumulation. Like aceruloplasminemia but distinct from most forms of NBIA, the pathophysiology of neuroferritinopathy explains the brain iron accumulation: the mutant protein disrupts the structure and iron-carrying capacity of ferritin, resulting in abnormal iron deposition in the brain.
Aceruloplasminemia
Aceruloplasminemia presents in adulthood and is characterized by microcytic anemia, diabetes, retinal disease, and a movement disorder consisting of facial dystonia, chorea, tremor, parkinsonism, ataxia, and cognitive decline [58,59]. In addition to anemia, laboratory investigations reveal low or absent serum ceruloplasmin, elevated ferritin, low iron, and low serum copper but normal urinary copper. Like neuroferritinopathy, it is a disorder of iron metabolism: mutations in the CP gene cause an absence of or reduction in the copper-carrying ceruloplasmin protein. This results in abnormal iron trafficking and deposition throughout the body, including the central nervous system. Iron chelation is routinely employed as a rational therapeutic, but while changes in iron deposition in peripheral organs and the brain can be demonstrated after several years of treatment, it is not clear whether this is accompanied by significant clinical improvement [60,61].
Woodhouse-Sakati syndrome
DCAF17 encodes a nucleolar protein thought to be involved in transcriptional regulation, and mutations in the gene result in an unusual syndrome of endocrine and neurologic abnormalities [62]. Affected individuals exhibit hypogonadism, diabetes mellitus, alopecia, abnormalities on electrocardiogram, extrapyramidal movement disorders, intellectual disability, and sensorineural healing loss [63,64]. MRI imaging reveals basal ganglia T2 hypointensities as well as white matter disease. Most of the described cases arise from a founder in the Saudi Arabian population, though affecteds of other ethnicities have been described [65].
Kufor-Rakeb syndrome
Kufor-Rakeb syndrome is a syndrome of juvenile-onset parkinsonism, spasticity, and cognitive decline originally described in a Jordanian family [66] and now described in multiple other ethnicities [67–71]. Variable clinical features include supranuclear gaze palsy, facial-finger-faucial mini myoclonus, and tremor. Generalized atrophy is a universal finding on brain MRI as the disease advances; basal ganglia iron may not be evident, especially early in disease. The parkinsonism is levodopa-responsive but, like MPAN and BPAN, management is complicated by the early development of motor fluctuations and dyskinesias. The disease is caused by mutations in the APT13A2 gene [72 encoding a lysosomal ATPase and is also known as PARK9.
CONCLUSIONS
The development of animal and cell-based model systems are advancing our understanding of the core pathophysiology of the disorders of brain iron accumulation, individually and collectively. Patient registries, natural history studies and well-curated biorepositories will be essential to support drug development efforts as rational therapeutic approaches to disease modification arise from the laboratory. Such efforts are underway.
Acknowledgments
We gratefully acknowledge the participation of NBIA patients and families in our research, and the support of or our work by the NBIA Alliance including the NBIA Disorders Association and Hoffnungsbaum e.V.
This publication was supported by Oregon Clinical and Translational Research Institute (OCTRI), grant number (UL1TR000128) from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Hogarth also receives funding support for NBIA research from the NBIA Disorders Association, the European Commission’s 7th Framework Programme (FP7/2007–2013, HEALTH-F2-2011, grant agreement No. 277984, TIRCON), and Retrophin, Inc. She receives funding support for work unrelated to NBIA from the Michael J. Fox Foundation, and Vertex, Inc.
Notes
Conflicts of Interest
The author has no financial conflicts of interest.