Dear Editor,
Cerebellar ataxia is a clinically and genetically heterogeneous disease. Several cerebellar ataxias are caused by genetic mutations with trinucleotide repeats, including spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, and 17 [1]. Some cerebellar ataxias are caused by single nucleotide substitutions that can be identified by next-generation sequencing (NGS), for example, whole-exome sequencing (WES). Among these, SCA42 is caused by pathogenic variants in the calcium channel 1G (CACNA1G) gene located on chromosome 17q21 [1], which encodes peptides consisting of the S4 segment of type channel protein Cav3.1 that is highly expressed in cerebellar tissue. Genetic mutations for this rare disease have been reported in Japanese, Chinese, Italian, European, and Yemeni families [1-4]. We recently encountered two Korean families with SCA42 associated with the known pathogenic mutation (NM_198384.3(CACNA1G): c.5144G>A, p.Arg1715His, heterozygote). To our knowledge, this is the first report of SCA42 with this mutation in South Korea.
Family 1 included a previously healthy 31-year-old Korean man who was referred to the neurology department for a progressive gait disturbance that began at age 25. He initially had trouble running and experienced insidiously progressive trouble while walking. He did not show any developmental delays in childhood, and his cognition was normal. A neurological examination showed that the patient was alert and oriented. The cranial nerve examination showed no abnormalities, including extraocular movement and saccades, and the sensory and motor function test findings were normal. Upper motor neuron signs, such as the Babinski reflex, ankle clonus, and hyperreflexia, were not observed. The patient’s gait was ataxic, with a broad base and irregular stepping, and he showed scanning speech, dysmetria, and dysdiadochokinesia in a cerebellar function test. His initial Scale for the Assessment and Rating of Ataxia (SARA) score was 9, and he could walk and perform daily activities without assistance. The patient’s history also showed restless legs since his twenties, which met the diagnostic criteria for restless leg syndrome (RLS). The RLS severity was 33 (out of 60) on the RLS-6 and 20 (out of 40) on the International Restless Legs Scale.
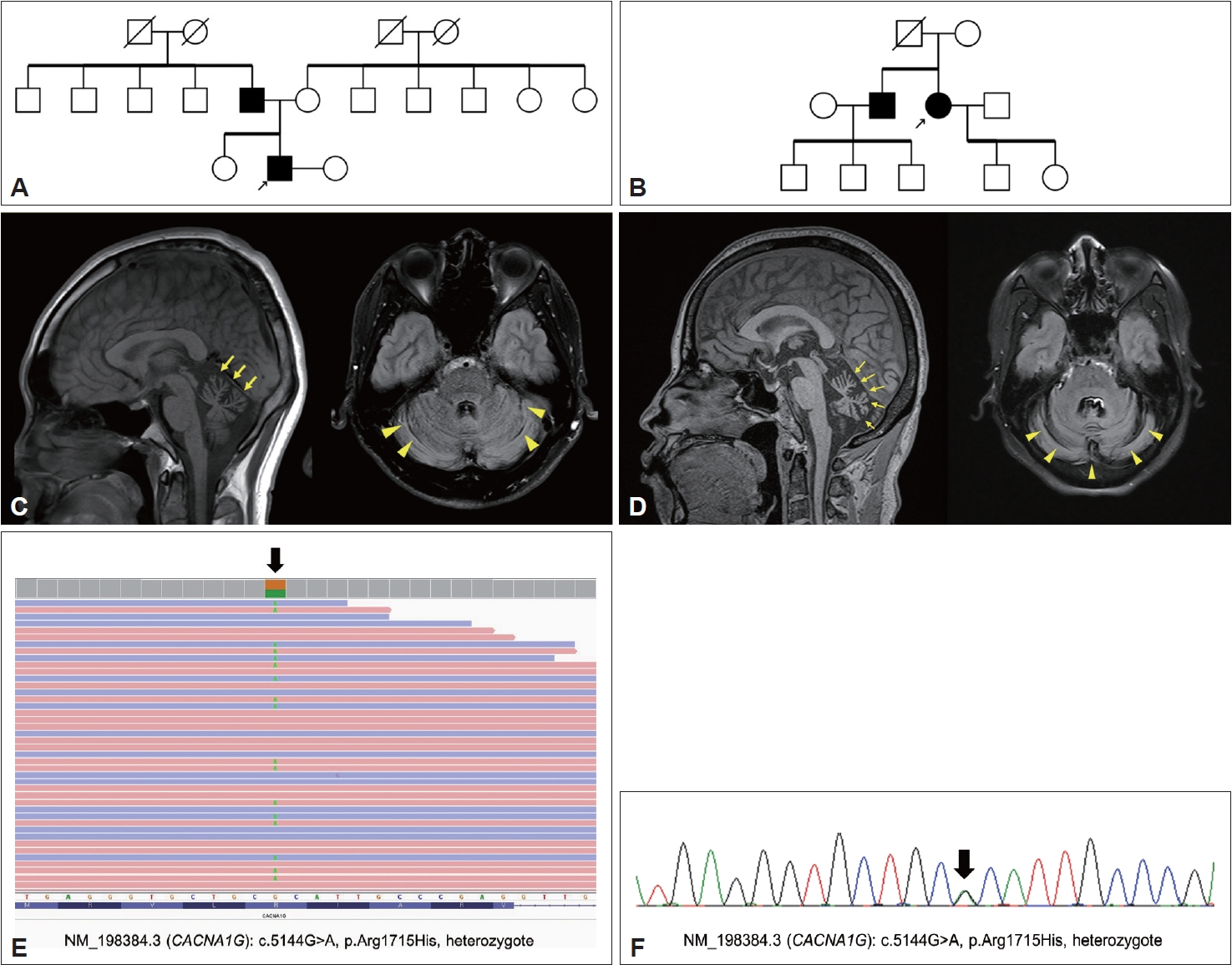
Examination of the family history revealed that the patient’s father was wheelchair-bound due to gait disturbance and imbalance beginning in his teenage years. All other family members were healthy. The three-generation family pedigree is shown in Figure 1A. Brain magnetic resonance imaging (MRI) of the patient showed cerebellar atrophy in both the vermis and cerebellar cortex (Figure 1C), without brainstem atrophy.
Highly suspicious of familial genetic etiology with an autosomal dominant pattern, we conducted genetic testing for SCA1, 2, 3, 6, 7, and 17, dentatorubral-pallidoluysian atrophy (DRPLA), in addition to an ataxia gene panel including 27 genes (ABCD1, APTX, ATL1(SPG3A), ATM, BEAN1, CACNA1A, CACNB4, CP, CYP27A1, FMR1, FXN, GFAP, KCNA1, MTTP, NPC1, PNKP, POLG, SACS, SETX, SLC1A3, SPAST(SPG4), SPG11, SPG7, SYNE1, TK2, TTPA, and ZFYVE26), all of which were negative. These findings prompted us to perform WES, which revealed a pathogenic variant causing SCA42, namely, NM_198384.3 (CACNA1G): c.5144G>A, p.R1715H (Figure 1E). The patient’s father showed an identical variant confirmed by Sanger sequencing (Figure 1F). The patient and his father were both diagnosed with SCA42.
Expecting the potential benefit of T-type calcium channel blockers, the patient was administered ethosuximide (up to 1,000 mg/day orally); however, the treatment was not effective. Another T-type calcium channel blocker, zonisamide, was administered orally up to 200 mg per day and did not improve ataxia symptoms. After increasing the dose of zonisamide, the patient experienced worsening RLS symptoms. After cessation of zonisamide treatment, the RLS symptoms returned to baseline levels.
Family 2 included a 55-year-old woman who visited the outpatient clinic of the neurology department for gait disturbances that began at age 48. The neurological examination showed scanning speech and ataxic gait. Slow and probable hypermetria were also observed. Neither abnormal nystagmus nor limitation on extraocular movement was observed. She did not show upper motor neuron signs, and her SARA scale was 12.5. She denied any symptoms of RLS. Examination of the family history revealed that one of her brothers was also affected with ataxic gait with dysarthria that began at age 40, and he did not show other oculomotor findings (Figure 1B). The patient’s father had died after being bedridden for 20 years due to a traffic accident in his 50s; however, she could not recall whether her parents had any symptoms of cerebellar ataxia. An MRI scan of her showed severe cerebellar atrophy in both the vermis and the cerebellar cortex (Figure 1D). Genetic testing was negative for SCA types 1, 2, 3, 6, 7, 8, 17, DRPLA, and the ataxia gene panel. Subsequent WES revealed a CACNA1G variant (c.5144G>A, p.Arg1715His). Sanger sequencing confirmed that the patient’s brother harbored the same pathogenic variant. We performed ancestry and relatedness analysis using the Peddy methodology [5] and validated that the cases were Korean (East Asian origin) but unrelated (Supplementary Figure 1 in the online-only Data Supplement). Ethosuximide (up to 500 mg/day) and zonisamide (up to 300 mg/day) were administered, with no significant improvement in her symptoms and signs.
This study is the first to identify SCA42 caused by the same pathogenic variant in CACNA1G in two unrelated Korean families. The clinical presentation of SCA42 includes insidiously progressive symptoms and signs as a mainly pure form of cerebellar ataxia [3]. CACNA1G encodes the T-type calcium channel Cav3.1, which is highly expressed in the molecular layer of the cerebellum, the inferior olive nucleus, and the thalamus and plays a role in regulating membrane potential and the calcium-mediated signaling pathway [6]. Among several mutations related to SCA42, the rare heterozygote mutation c.5144G>A, p.Arg1715His, located in the S4 segment of Cav3.1, has been reported in French [3], Italian, Yemeni [4], and Japanese [1,2] families (Supplementary Table 1 in the online-only Data Supplement), and it is classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines. Arginine is highly conserved in this channel, and variants in this arginine in the S4 segment disrupt its voltage-sensing function and electrophysiological features of the channel [1]. Ongoing case reports and studies aim to identify the detailed pathophysiology related to the genotype and phenotype of SCA42. In the case of early onset severe SCA42 with neurodevelopmental deficits (SCA42ND), pathogenic variants in the CACNA1G gene involve p.Ala961Thr, p.Met1531Val, and p.Ile1273Phe. These variants were demonstrated to cause significant changes in T-type Ca2+ currents in electrophysiological studies using HEK293T cells. However, a genotype-phenotype correlation has not yet been established for the CACNA1G gene, and further studies are needed.
Our patients had clinical presentations compatible with those previously reported in families with SCA42. Interestingly, our patient in Family 1 complained of restless legs, which met the diagnostic criteria for RLS. While the relationship between RLS and SCA42 has not been previously reported, patients with SCA1, SCA2, and SCA3 are known to be more susceptible to RLS than unaffected controls. The results of recent positron-emission tomography and genetic studies suggest the role of the dopaminergic pathway as a possible shared pathophysiology for patients with RLS and SCA. Because the RLS symptoms worsened after the administration of zonisamide in the patient of Family 1 and several other reports [7] and medications to control RLS symptoms involve the alpha-2-delta calcium channel ligands gabapentin and pregabalin, calcium channels may be involved in the pathophysiology of RLS. Although the evidence does not yet exist, future studies might be considered for the possible common pathophysiology related to the calcium channel pathway of RLS and SCA42.
Ataxia in adults can be difficult to diagnose because of heterogeneity in causative genetic mutations; therefore, many patients remain undiagnosed. To date, more than 40 genetically distinct types of SCA have been identified. Tandem repeat expansion is the major genetic cause of SCA and can be detected by polymerase chain reaction (PCR)-based testing. However, recent reports have revealed various types of SCA caused by sequence variants, including SCA13, 19, 21, 29, 41, 42, and 44. This justifies the need for NGS testing in patients with undiagnosed ataxia. Recently, the first Korean cases of SCA13, 19, and 21 were diagnosed by NGS in patients within single families. Our identification of patients carrying the same pathogenic variant from two unrelated families is remarkable, as the CACNA1G c.5144G>A mutation has also been reported in Japan [1], and it is likely to be a founder mutation in East Asia, including Korea. Therefore, SCA42 should be suspected as an important cause of undiagnosed ataxia in Korea that has not yet been identified due to insufficient genetic testing.
In summary, we identified the first Korean cases of SCA42 in two unrelated families. This report highlights the importance of NGS testing in Korean patients with ataxia who remain undiagnosed after testing for tandem repeat disorders. As the prevalence of SCA42 in each ethnicity or country is unknown, further studies are warranted to unravel the genetic characteristics of this disease.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.22150.
Supplementary Table 1.
Literature review of SCA42 cases/families reported with the missense mutation (c.5144G>A, p.R1715H) in the CACNA1G gene
jmd-22150-Supplementary-Table-1.pdf
Supplementary Figure 1.
Ancestry and pair-wise relatedness analysis using Peddy in our cases. A: Ancestry analysis of the two cases. We used exome sequencing data and the 1000 Genomes Project reference panel (1KGP) to validate both cases are Koreans (East Asian ancestry). B: Pair-wise comparison of two cases confirmed that both cases are unrelated.
jmd-22150-Supplementary-Figure-1.pdf
Notes
-
Ethics Statement
Written consent was obtained from patients for the publication.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This research was supported by a fund (2020-ER6904-01) by Research of Korea Centers for Disease Control and Prevention.
-
Author Contributions
Conceptualization: Jangsup Moon, Han-Joon Kim. Investigation: all authors. Supervision: Jangsup Moon, Han-Joon Kim. Visualization: Hyoshin Son, Jihoon G. Yoon, Man Jin Kim. Writing—original draft: Hyoshin Son. Writing—review & editing: all authors.
Figure 1.The pedigrees of two families, brain magnetic resonance imaging (MRI), and the genetic results of two unrelated Korean families with SCA42. A: Pedigree of Family 1. The proband marked with the arrow and his father were symptomatic for cerebellar ataxia. B: Pedigree of Family 2. Arrow: proband of Family 2. The proband’s father was bedridden after a traffic accident in his 50s and is deceased. Except for the patient and her brother, no other family members showed neurological symptoms or signs. Genetic tests of the asymptomatic members were not performed according to their decisions. The proband and her brother appeared to have the same genetic mutation (NM_198384.3(CACNA1G): c.5144G>A, p.Arg1715His, heterozygote). C: Cerebellar atrophy in the sagittal (left, yellow arrow) and axial (right, arrowhead) MRI views of the proband of Family 1. D: Severe cerebellar atrophy in the sagittal (left, yellow arrow) and axial (right, arrowhead) views of the MRI of the proband of Family 2. E: Exome sequencing results of the proband of Family 1 showing a genetic mutation: NM_198384.3(CACNA1G): c.5144G>A, p.Arg1715His, heterozygote. F: Genetic mutation (black arrow) of the patient’s father confirmed by Sanger sequencing (NM_198384.3(CACNA1G): c.5144G>A, p.Arg1715His, heterozygote).
REFERENCES
- 1. Morino H, Matsuda Y, Muguruma K, Miyamoto R, Ohsawa R, Ohtake T, et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain 2015;8:89.ArticlePubMedPMCPDF
- 2. Kimura M, Yabe I, Hama Y, Eguchi K, Ura S, Tsuzaka K, et al. SCA42 mutation analysis in a case series of Japanese patients with spinocerebellar ataxia. J Hum Genet 2017;62:857–859.ArticlePubMedPDF
- 3. Coutelier M, Blesneac I, Monteil A, Monin ML, Ando K, Mundwiller E, et al. A recurrent mutation in CACNA1G alters Cav3.1 T-type calciumchannel conduction and causes autosomal-dominant cerebellar ataxia. Am J Hum Genet 2015;97:726–737.ArticlePubMedPMC
- 4. Ngo K, Aker M, Petty LE, Chen J, Cavalcanti F, Nelson AB, et al. Expanding the global prevalence of spinocerebellar ataxia type 42. Neurol Genet 2018;4:e232.ArticlePubMedPMC
- 5. Pedersen BS, Quinlan AR. Who’s who? Detecting and resolving sample anomalies in human DNA sequencing studies with peddy. Am J Hum Genet 2017;100:406–413.ArticlePubMedPMC
- 6. Hara N, Morino H, Matsuda Y, Satoh K, Hashimoto K, Maruyama H, et al. Zonisamide can ameliorate the voltage-dependence alteration of the T-type calcium channel CaV3.1 caused by a mutation responsible for spinocerebellar ataxia. Mol Brain 2020;13:163.ArticlePubMedPMCPDF
- 7. Chen JT, Garcia PA, Alldredge BK. Zonisamide-induced restless legs syndrome. Neurology 2003;60:147.ArticlePubMed
Citations
Citations to this article as recorded by

- Targeting Ion Channels and Purkinje Neuron Intrinsic Membrane Excitability as a Therapeutic Strategy for Cerebellar Ataxia
Haoran Huang, Vikram G. Shakkottai
Life.2023; 13(6): 1350. CrossRef
 E-submission
E-submission
 , Jihoon G. Yoon2
, Jihoon G. Yoon2

 PubReader
PubReader ePub Link
ePub Link Cite
Cite
