Department of Neurology, National Institute of Mental Health & Neurosciences, Karnataka, India
Corresponding author: Pramod Kumar Pal, MD, DNB, DM, FRCP Department of Neurology, National Institute of Mental Health & Neurosciences, Hosur Road, Bangalore, Karnataka 560029, India / Tel: +91-80-26995147 / Fax: +91-80-26564830 / E-mail: palpramod@hotmail.com
٭These authors contributed equally to this work.
• Received: July 4, 2022 • Revised: September 16, 2022 • Accepted: October 29, 2022
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
GTP cyclohydrolase-1 (GCH1)-deficient dystonia (DYT-GCH1/DYT5) is an autosomal-dominant dopa-responsive dystonia (DRD) of childhood onset typically presenting with foot dystonia progressing to generalized dystonia and parkinsonism with diurnal fluctuation and a dramatic and persistent response to levodopa (LD) [1]. In addition, nonmotor symptoms such as depression, anxiety and sleep quality impairment have been reported and can predate motor symptom onset [2]. Occasionally, the phenotypic spectrum may include upper-limb (UL) onset of dystonia, focal dystonia, and adult-onset parkinsonism and may mimic cerebral palsy or spastic paraplegia. Apart from these, there can be additional atypical features (DRD-plus), such as infantile onset, developmental delay, hypotonia, ptosis, cerebellar dysfunction, and poor LD responsiveness [3]. We report one such case of DRD-plus presentation of DYT-GCH1 with myoclonus dystonia and associated neuropsychiatric symptoms wrongly diagnosed as functional movement disorder at onset.
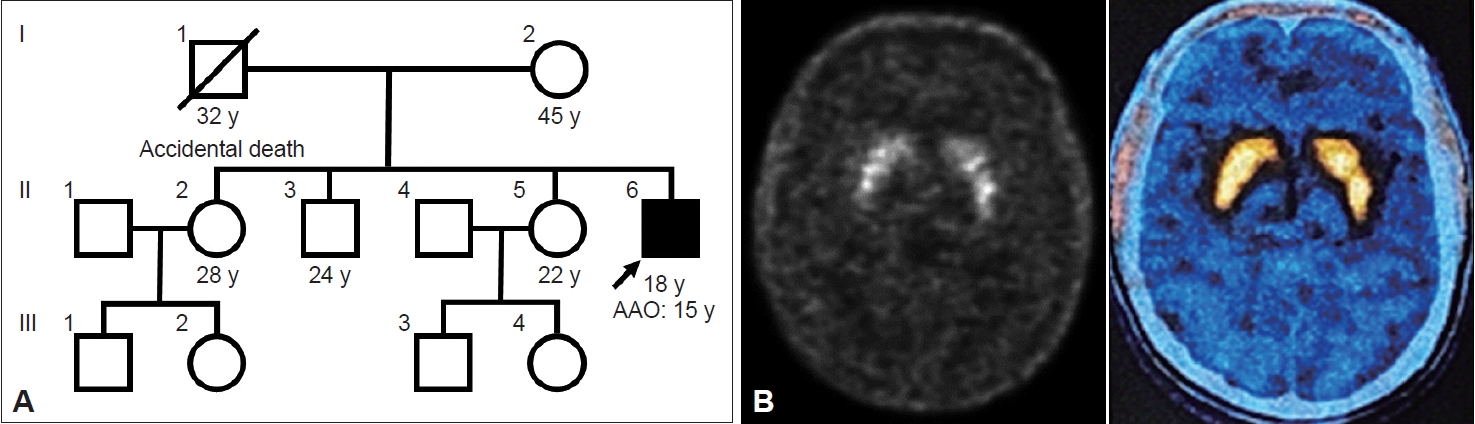
An 18-year-old male, teetotaler with normal perinatal, developmental and family history (Figure 1A), presented with a three-year history of jerky involuntary movements of the proximal ULs, which were intermittent at onset and later became persistent after a year. He also had jerky backward movements of the head with difficulty speaking and swallowing. The UL movements occurred even at rest, while movements of the head worsened upon sitting and speaking. Sleep benefit was noted, which lasted approximately 15–20 minutes without any obvious diurnal variation. The patient apparently frustrated by these movements, previously harmed himself using sharp objects. In addition, one year before symptom onset, the patient had behavioral changes including a reduced interest in studies, a decline in scholastic performance, obsessive-compulsion and increased anger outbursts. He consulted several doctors regarding these behavioral changes, and the possibility of functional movement disorder was considered. There was no response to clonazepam, trihexyphenidyl, primidone or propranolol. The movements were reduced with tetrabenazine; however, he developed significant slowness of all activities during which he first consulted us. The patient had symmetrical parkinsonism with bilateral UL resting and postural tremors. Based on the history and videos that the patient provided, a provisional diagnosis of myoclonus dystonia with drug-induced parkinsonism was considered. He was advised to stop tetrabenazine and assess when the movements reappeared. After 10 days, all the signs of parkinsonism had disappeared, and the initial abnormal movements had reappeared. Informed consent was obtained from the patient for video recording and publication.
On examination, cognition was normal with a Montreal Cognitive Assessment score of 28, and normal fundus, eye movements and speech. He had healed scars from self-inflicted wounds over the forearms, upper arms and back. There was intermittent blepharospasm, facial grimacing with frequent jerky movements associated with posturing of both the ULs, head and occasionally the trunk (Supplementary Video 1, segment 1 in the online-only Data Supplement), suggesting myoclonus-dystonia. These movements could be suppressed voluntarily for a few seconds, but there was no associated urge or rebound to suggest tics. He had bilateral UL postural-jerky tremors that persisted in action but not at rest. The gait was normal except for the pronounced head and UL movements. The rest of the examination was normal. The patient’s brother and mother were examined, and their clinical assessments were normal.
Based on the examination findings, the possibility of adolescent-onset, progressive, persistent, generalized combined dystonia with myoclonus was considered. Biochemical blood investigations, magnetic resonance imaging of the brain and Flourodopa positron emission tomography-computed tomography (Figure 1B) performed in view of tetrabenazine-induced parkinsonism were normal. Whole-exome sequencing revealed a novel heterozygous missense likely pathogenic mutation in the GCH1 gene (NM_001024024.1:c.449G>A;p.Gly150Glu) (Supplementary Material in the online-only Data Supplement). A diagnosis of myoclonus-dystonic presentation of DYT-GCH1 was made. After one week of 400 mg/100 mg/day of LD/carbidopa (CD), the patient had significant improvement (Supplementary Video 1, segment 2 and 3 in the online-only Data Supplement) in both movement (37.5 to 6.5; 82.7%) and disability (7 to 2; 71.4%) scores on the Burke-Fahn-Marsden dystonia rating scale (BFMDRS). The improvement was sustained at the one-month follow-up.
Inherited myoclonus-dystonia is a genetically heterogenous group with approximately one-third of cases accounted for by a mutation in the epsilon-sarcoglycan (SCGE) gene (DYT-SCGE, DYT11), an autosomal-dominant dystonia with “lightning-jerks” and dystonia predominantly affecting the neck and ULs with onset in the first to second decade. Some of the SCGE-negative myoclonus dystonia cases are due to other genes associated with dystonia, such as TOR1A, THAP1, ANO3, GNAL, CACNA1B, TITF1, ADCY5, KCTD1, RELN, and KCNN2 [4,5]. Rarely, mutations in genes associated with dopamine synthesis (GCH1, TH, SR, AADC) can also have myoclonus-dystonia manifestations [6]. Even though it is difficult to differentiate SCGE-positive and SCGE-negative myoclonus dystonia, the presence of truncal dystonia and the coexistence of myoclonus and dystonia in the same body part with action suggest an alternative cause other than DYT-SCGE [7]. Coexistent myoclonus and dystonia in the same body part as seen in our case may suggest a “jerky-dystonic” instead of myoclonus. Moreover, the jerks in DYT-SCGE are often described as “lightening” compared to the slower jerks in other cases, including ours.
To the best of our knowledge, there is a single report of myoclonus-dystonia presentation of DYT-GCH1. Leuzzi, et al. [3] described a 17-year-old male with childhood onset myoclonus with UL onset later involving the lower limbs, face and trunk, predominant action, associated with mild dystonia of ULs and neck, mild bradykinesia, and lack of facial expression. A prompt, sustained but partial response was noted to 200 mg/50 mg/day of LD/CD. The patient carried a previously reported pathogenic heterozygous missense variant (c.671A>G;Lys22Arg) in GCH1 that segregated with the affected family members.
Nonmotor symptoms, predominantly neuropsychiatric, such as depression, anxiety and obsessive-compulsive disorders, are well reported in patients with inherited dystonia, especially the DRD spectrum, including DYT-GCH1, and can even predate motor symptom onset [2]. Impairment in monoamine, catecholamines and serotonin synthesis with mutations in DRD-spectrum genes may be the underlying mechanism for the range of nonmotor symptoms. The onset of psychiatric symptoms, especially depression and anxiety followed by movement disorders, may sometimes lead to a wrong diagnosis of functional movement disorder; thus, may lead to an avoidable delay in investigations and adequate therapy to improve the quality of life. It may also lead to exposure to unwarranted psychiatric medications, which often lead to de novo movement disorders such as tetrabenazine-induced parkinsonism, as seen in our case, or worsening of the underlying movement disorders.
To conclude, DYT-GCH1 should be considered in patients with inherited myoclonus-dystonia. Neuropsychiatric symptoms are well documented in DRD, including DYT-GCH1, and can predate motor symptoms. Lack of awareness of these nonmotor symptoms can sometimes lead to an erroneous diagnosis of functional movement disorder.
Pre- and post-treatment videos of the patient. Segment 1: Video demonstrating normal speech, cranial and proximal upper limb predominant dystonia with myoclonus characterized by adduction and internal rotation at the shoulders with extension at the elbow and flexion at the wrist and fingers. There was intermittent backward jerking of the neck with anterior and downward movements of the chin, indicating retrocollis with antecaput, facial dystonia and backward extension of the upper part of the trunk. Segment 2: Significant improvement in the severity of the phenomenology after 1 week of 400 mg/100 mg/day of levodopa/carbidopa. Segment 3: Pre- and post-treatment videos side-by-side to demonstrate the improvement.
SUPPLEMENTARY MATERIAL
DETAILS OF THE IDENTIFIED GENETIC VARIANT IN THE PATIENT
The authors confirm that the approval from an institutional review board of National Institute of Mental Health & Neurosciences was obtained for this work (No. NIMHANS/24th IEC (BS & NS DIV.)/2020 dated 25-06-2020). We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. We also confirm that the patient has given written informed consent for the publication of his video.
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
Parkinson’s Disease and Movement Disorders Research Fund and Indian Council of Medical Research (No. 54/3/2020-HUM/BMS).
Author contributions
Conceptualization: Praveen Sharma, Vikram V Holla, Pramod Kumar Pal. Data curation: Praveen Sharma, Vikram V Holla, Sandeep Gurram. Formal analysis: Praveen Sharma, Vikram V Holla. Pramod Kumar Pal. Funding acquisition: Vikram V Holla, Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Investigation: Praveen Sharma, Vikram V Holla, Nitish Kamble, Pramod Kumar Pal. Project administration: Nitish Kamble. Supervision: Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Validation: Nitish Kamble, Ravi Yadav, Pramod Kumar Pal. Visualization: Praveen Sharma, Vikram V Holla, Pramod Kumar Pal. Writing—original draft: Praveen Sharma. Writing—review & editing: all authors.
Figure 1.
Pedigree and molecular imaging of the patient. A: Pedigree of the patient. B: Flouro-DOPA PET-CT of the patient showing bilateral symmetrical uptake in the basal ganglia. AAO, age at onset; PET-CT, positron emission tomography-computed tomography; y, years.
REFERENCES
1. Furukawa Y. GTP cyclohydrolase 1-deficient dopa-responsive dystonia. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(R). Seattle: University of Washington; 2019.
2. Antelmi E, Stamelou M, Liguori R, Bhatia KP. Nonmotor symptoms in dopa-responsive dystonia. Mov Disord Clin Pract 2015;2:347–356.ArticlePubMedPMCPDF
3. Leuzzi V, Carducci C, Carducci C, Cardona F, Artiola C, Antonozzi I. Autosomal dominant GTP-CH deficiency presenting as a dopa-responsive myoclonus-dystonia syndrome. Neurology 2002;59:1241–1243.ArticlePubMed
4. Lange LM, Junker J, Loens S, Baumann H, Olschewski L, Schaake S, et al. Genotype–phenotype relations for isolated dystonia genes: MDSGene systematic review. Mov Disord 2021;36:1086–1103.ArticlePubMedPDF
5. Balint B, Bhatia KP. Isolated and combined dystonia syndromes - an update on new genes and their phenotypes. Eur J Neurol 2015;22:610–617.ArticlePubMed
6. Stamelou M, Mencacci NE, Cordivari C, Batla A, Wood NW, Houlden H, et al. Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency. Neurology 2012;79:435–441.ArticlePubMedPMC
7. Zutt R, Dijk JM, Peall KJ, Speelman H, Dreissen YE, Contarino MF, et al. Distribution and coexistence of myoclonus and dystonia as clinical predictors of SGCE mutation status: a pilot study. Front Neurol 2016;7:72.ArticlePubMedPMC
Figure & Data
References
Citations
Citations to this article as recorded by
A Genetics Pearl for Counseling Patients with Epsilon-Sarcoglycan Myoclonus-Dystonia Alissa S. Higinbotham, Suzanne D. DeBrosse, Camilla W. Kilbane Tremor and Other Hyperkinetic Movements.2023;[Epub] CrossRef
 E-submission
E-submission
 , Vikram V Holla*
, Vikram V Holla*


 PubReader
PubReader ePub Link
ePub Link Cite
Cite