11st Department of Neurology, AHEPA University Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
2Computational Clinical Imaging Group, Centre for the Unknown, Champalimaud Foundation, Lisbon, Portugal; Royal Marsden, London, UK; The Institute of Cancer Research, London, UK; Karolinska Institute, Sweden; Institute of Computer Science, The Foundation for Research andTechnology – Hellas (FORTH), Heraklion, Greece
3Interbalkan Medical Center of Thessaloniki, Thessaloniki, Greece; Department of Radiology, Aristotle University of Thessaloniki, Thessaloniki, Greece
43rd Department of Neurology, “G.Papanikolaou” Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
Corresponding author: Vasilios K. Kimiskidis, MD, PhD 1st Department of Neurology, AHEPA University Hospital, Aristotle University of Thessaloniki, 1 Stilp Kyriakidi str, 54621, Thessaloniki, Greece / Tel: +302310994667 / Fax: +302310994670 / E-mail: kimiskid@auth.gr
• Received: January 28, 2022 • Revised: April 12, 2022 • Accepted: April 28, 2022
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Paroxysmal kinesigenic dyskinesia (PKD) is an inherited or acquired movement disorder that may occur in patients with multiple sclerosis (MS) [1]. Secondary paroxysmal dyskinesia (SPD), a rare manifestation of MS, has a prevalence that ranges from 0.16% to 1.53% in different studies [1,2] and may even occur as a presenting symptom of MS. The therapeutic mainstay includes steroids and antiepileptic drugs [1,2]. We describe a patient presenting with secondary PKD due to an MS-related demyelinating lesion in the right cerebral peduncle. The patient failed to respond to steroids and levetiracetam but showed rapid improvement with lacosamide (LCM).
A 35-year-old female presented to the emergency department of our hospital with a new-onset movement disorder. The patient experienced three brief episodes of tingling in the left part of her body followed by painful cramps in the past 24 hours. During the attacks, her left foot and hand were held in a dystonic flexed position (internal rotation of the foot and flexion of the wrist and fingers), and there was forceful contraction of her mouth to the left (Supplementary Video 1 in the online-only Data Supplement). These episodes lasted approximately 30 seconds and were triggered by sudden changes in movement, e.g., when getting up from a chair. The patient’s level of consciousness remained intact during the episodes. Her past medical history was notable for endoscopic transsphenoidal surgery for pituitary adenoma 8 years ago. Her current medications include desmopressin for central diabetes insipidus and levothyroxine. There was no other relevant personal or familial history.
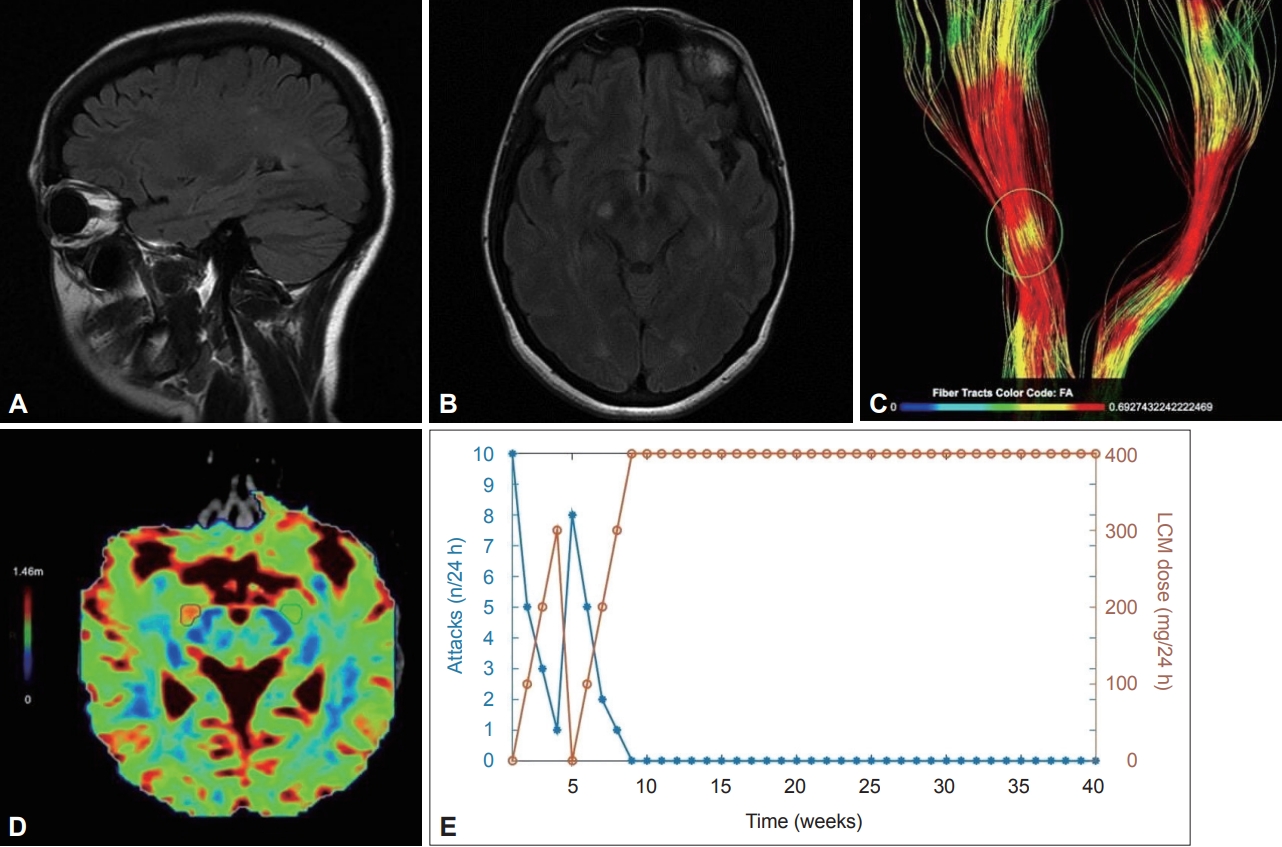
The neurological examination as well as blood hematology and biochemistry tests were normal. The electroencephalogram was also normal. Brain MRI showed small, scattered hyperintensities in the periventricular and subcortical white matter in Τ2-weighted and FLAIR sequences. In addition, a 7 mm hyperintense contrast-enhancing lesion in the right cerebral peduncle was revealed and thought to be relevant for the patient’s symptoms (Figure 1A and B). In order to investigate the microstructural integrity of the corticospinal tract and explore the microstructural alterations that may be responsible for the patient’s attacks, MR tractography was performed and revealed an area of pathology that corresponded topographically to the midbrain lesion (Figure 1C and D). Cerebrospinal fluid (CSF) analysis showed mild lymphocytic pleocytosis (5–6 cells/μL) and an elevated IgG index with positive oligoclonal bands. Viral PCR tests of CSF and immunological testing for autoimmune encephalitis, including anti-leucine-rich glioma-inactivated 1 (LGI1) and anti-N-methyl-D-aspartate (NMDA) receptor antibodies, were negative. Aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) antibodies were also negative. The patient’s diagnosis was MS according to the 2017 McDonald criteria. A genetic test for familial PKDs was not performed because of the patient’s age and negative family history.
During her hospitalization, the patient experienced up to ten attacks daily. Intravenous steroids (methylprednisolone 1 g/24 h for 5 days) and levetiracetam (3 g/24 h) produced no response. Since the patient was receiving desmopressin, carbamazepine was avoided because its combined use with desmopressin might increase the risk of adverse effects (i.e., hyponatremia). Prompted by the positive experience with LCM in four cases of idiopathic PKDs and in one case of SPD attributed to neuromyelitis optica spectrum disorder (NMOSD) [3,4], LCM (gradually up-titrated to 300 mg/24 h per os) was administered and significantly reduced the frequency of the attacks (Figure 1E). Five weeks later, LCM was discontinued, which led to a severe relapse. The reinitiation of LCM (400 mg/24 h) resulted in complete and long-lasting attack cessation. Glatiramer acetate 40 mg TPW was also initiated as a first-line treatment for MS. Follow-up MRIs of the brain and cervical spine at three and six months showed a stable T2 lesion load.
The pathophysiology of PKD in MS remains uncertain. Ostermann and Westerberg [5] proposed that paroxysmal dystonia in MS patients is likely caused by an ephaptic (nonsynaptic) activation of corticospinal axons within a demyelinating plaque at any level in the motor pathway. The location of the lesion, in our case, is rather unusual, as evidenced by a voxel-based lesion symptom mapping study that revealed prominent associations between SPD and MS lesions in the internal capsule, basal ganglia and posterior thalamic radiation [2]. Another possible mechanism is ion channel dysfunction in partially demyelinated axons. To support saltatory conduction, normal myelinated axons exhibit an aggregation of sodium channels in the axon membrane at the nodes of Ranvier, with a much lower density in internodal domains. Following demyelination, the expression of voltage-gated sodium channels is increased in the axonal membrane, which promotes the recovery of action potential conduction [6]. Transient ion channel dysfunction in the setting of an active demyelinating lesion was proposed as a pathogenetic mechanism because an inflammatory milieu may predispose neuronal channel function toward hyperexcitability [6].
LCM has a multipathway mechanism of action, including the selective enhancement of the slow inactivation of voltage-gated sodium channels, which stabilizes hyperexcitable neuronal membranes and blocks neuronal firing [7]. This mechanism differs from carbamazepine, which primarily enhances the fast inactivation of voltage-gated sodium channels.
In line with previous reports, LCM in the presented case provided prompt and complete PKD control [3,4]. In contrast, however, to a case series of 7 MS patients [1] where SPD did not recur after 4 weeks of treatment, the discontinuation of LCM led to a severe relapse which responded to drug reinitiation. This observation suggests that SPD in MS patients may, on occasion, require long-term treatment. The effectiveness of LCM in the symptomatic management of tonic spasms related to transverse myelitis in the context of NMOSD [4] indicates that LCM can be a useful therapeutic option for SPD related to demyelinating diseases other than MS. LCM was initiated following the diagnosis of carbamazepine-induced Stevens-Johnson syndrome in the patient with NMOSD, which was contrary to the initiation of LCM as a first-choice drug in our patient.
In conclusion, LCM may be a useful therapeutic option for patients with MS-related PKD because of its mechanism of action and its fast, significant and sustained efficacy. Although additional evidence is needed to draw a definitive conclusion about the efficacy of LCM for PKD, its favorable tolerability profile compared to carbamazepine may prompt clinicians to investigate its use as a first-choice drug in patients with secondary or idiopathic PKDs.
Notes
Ethics Statement
All procedures performed in this study were conducted in accordance with the ethical standards of the institutional and/or national research committee and with the 1975 Helsinki declaration and its later amendments or comparable ethical standards. The patient gave informed consent, allowing publication of the clinical data and the video.
Supplementary Video Legends
Video 1.
The video demonstrates a typical attack in which the patient’s left hand was held in a dystonic flexed position. This episode lasted approximately 30 seconds and was triggered by a sudden change in movement when the patient got up from the chair. Neither hyperventilation nor other factors, such as caffeine or stress, precipitated the attack.
Maria Moschou received research support from Sanofi Genzyme, Genesis and UCB. Vasilios K. Kimiskidis received funding and/or honoraria from Arriani, Bial, Eisai, Genesis, Janssen-Cilag, Merck Serono, Novartis, Teva and UCB.
All remaining authors have declared no conflicts of interest.
Funding Statement
None
Author Contributions
Data curation: Nickolas Papanikolaou, Antonios Drevelegas. Supervision: Martha Spilioti, Vasilios K. Kimiskidis. Writing—original draft: Vasiliki Poulidou. Writing—review & editing: all authors.
Acknowledgments
We would like to thank Antonis Frontistis for editing the video.
Figure 1.
Brain MRI examination was performed on a 3T system (Siemens Magnetom Vida 3T, Siemens Healthineers, Erlangen, Germany) using a 64-channel head coil. Anatomical imaging based on a 3D T1-weighted sequence was acquired using an MPRAGE sequence (voxel-size 1 × 1 × 1 mm, sagittal slice, orientation, matrix size 240 × 240). DTI acquisition included an axial single-shot spin-echo echoplanar imaging sequence with 32 diffusion encoding directions (field of view: 240 × 240 mm, acquisition voxel size: 2 × 2 × 2 mm, sensitivity encoding reduction factor of 2, multislice acceleration factor of 2, two b factors with 0 s/mm2 (low b), and 1,000 s/mm2 (high b), scan time was 2 min 36 s). DTI tractography analysis comprised the examination of white matter tracts as 3-dimensional and quantitative data of white matter fibers (corticospinal tract, inferior fronto-occipital fasciculus, corpus callosum). The Brainance DTI suite (Advantis Medical Imaging, Eindhoven, The Netherlands) was used for the reconstruction and visualization of 3-dimensional white matter tracts and the extraction of quantitative data (apparent diffusion coefficient, fractional anisotropy, radial diffusivity). A and B: Axial and sagittal FLAIR MRI scans of the brain show a hyperintense lesion in the right cerebral peduncle along with scattered hyperintensities in the subcortical and periventricular white matter, primarily in the vicinity of the trigones of the lateral ventricles. C: Reconstruction of the corticospinal tracts with color encoding based on the variations in FA. The area of the pathology is highlighted, and a reduction in FA is observed related to disorganized tracts. D: Color encoded map of radial diffusivity. The area with high signal intensity is mapped with red color visualizing the extension of the total area close to the lesion with a 20.7% increase in radial diffusivity compared to the green marked contralateral region of interest with normal values. Radial diffusivity reflects diffusivity perpendicular to axonal fibers and appears to be more strongly correlated with myelin abnormalities, either dysmyelination or demyelination. E: The relationship between LCM dose and the frequency of the attacks. Significant remission of the attacks was achieved within 3 days of initiating low-dose LCM, which was subsequently uptitrated to 300 mg daily, leading to almost complete cessation of the attacks. After an episode-free period, LCM was abruptly discontinued, leading to recurrence of the attacks (up to eight per day), thereby proving that the declining frequency of the attacks in the first four weeks was casually linked to LCM rather than to the natural course of PKD. Ten months later, the patient remained free of attacks with 400 mg LCM daily. DTI, diffusion tensor imaging; FA, fractional anisotropy; LCM, lacosamide; PKD, paroxysmal kinesigenic dyskinesia.
REFERENCES
1. Ciampi E, Uribe-San-Martín R, Godoy-Santín J, Cruz JP, Cárcamo-Rodríguez C, Juri C. Secondary paroxysmal dyskinesia in multiple sclerosis: clinical-radiological features and treatment. Case report of seven patients. Mult Scler 2017;23:1791–1795.ArticlePubMed
2. Fröhlich K, Winder K, Linker RA, Huhn K, Engelhorn T, Dörfler A, et al. Lesion correlates of secondary paroxysmal dyskinesia in multiple sclerosis. J Neurol 2018;265:2277–2283.ArticlePubMed
3. Furukawa G, Negishi Y, Takeuchi T, Ishihara N, Okumura A. Lacosamide for children with paroxysmal kinesigenic dyskinesia. Brain Dev 2020;42:617–620.ArticlePubMed
4. Baheerathan A, Brownlee WJ, Rugg-Gunn F, Chard DT, Trip SA. Neuromyelitis optica spectrum disorder related tonic spasms responsive to lacosamide. Mult Scler Relat Disord 2017;13:73–74.ArticlePubMed
6. Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 2006;7:932–941.ArticlePubMed
7. Carona A, Bicker J, Silva R, Fonseca C, Falcão A, Fortuna A. Pharmacology of lacosamide: from its molecular mechanisms and pharmacokinetics to future therapeutic applications. Life Sci 2021;275:119342.ArticlePubMed
 E-submission
E-submission
 , Martha Spilioti1
, Martha Spilioti1

 PubReader
PubReader ePub Link
ePub Link Cite
Cite