Dear Editor,
Elevated serum alpha-fetoprotein (AFP) can be an important clue for the diagnosis of certain types of autosomal recessive cerebellar ataxias, including ataxia telangiectasia (AT) and ataxia with oculomotor apraxia type 2 (AOA2), AOA1, or AOA4. In these disorders, diagnosis is confirmed through genetic testing based on clinical characteristics. However, confirmatory diagnosis is challenging when only a heterozygote mutation is found in conventional genetic testing. Recently, with the development of genetic technology, copy number variation (CNV) analysis has become available for these disorders. Here, we report a case of AOA2 in which a clinically suspected diagnosis was genetically confirmed through CNV analysis.
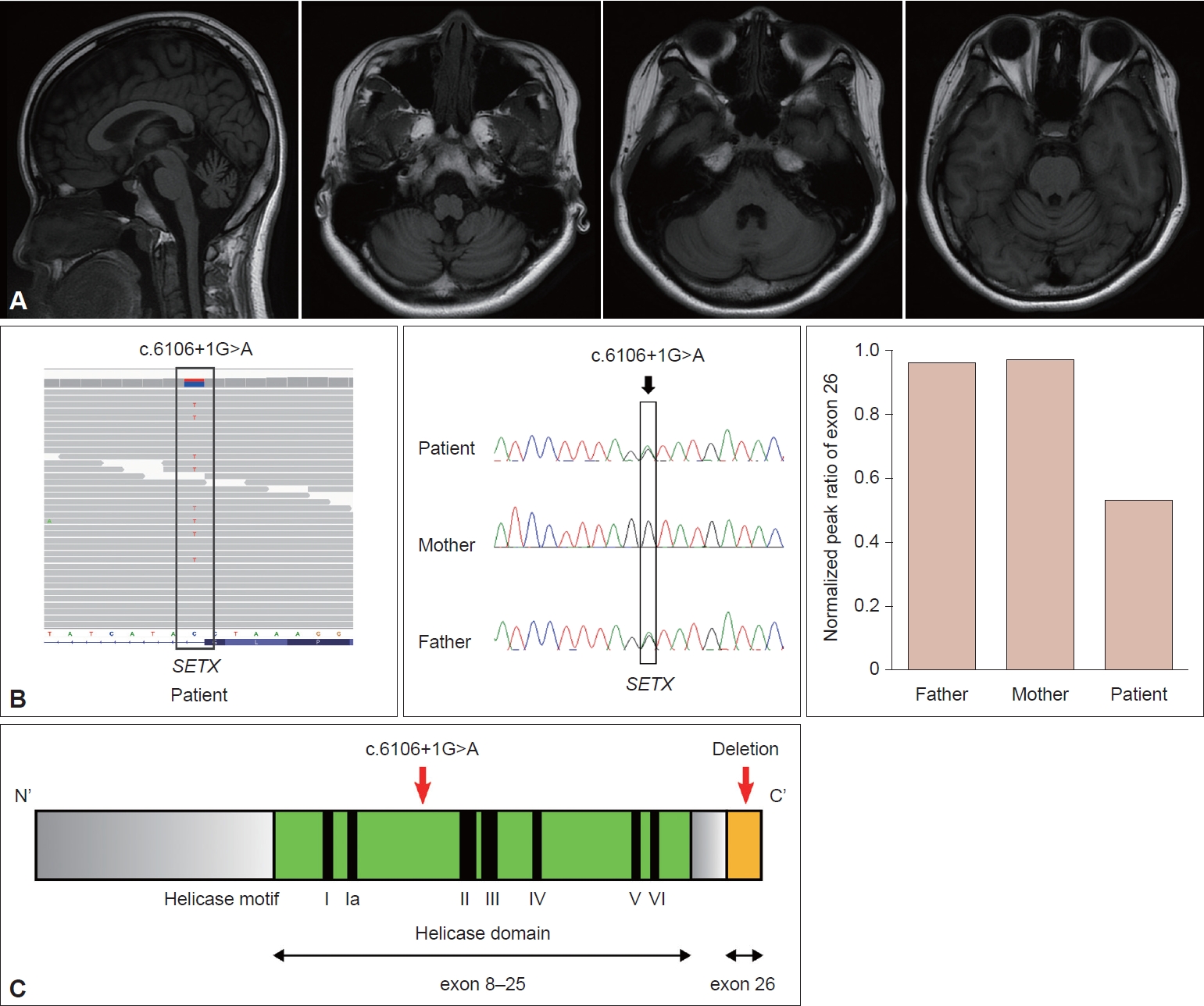
An 18-year-old female patient was referred due to progressive gait disturbance that started three years prior. She had normal developmental milestones and no cognitive impairment. The family history was unremarkable. On neurological examination, cerebellar dysfunction was noted, including truncal and limb ataxia, dysarthria, and dysdiadochokinesia. Oculomotor examination revealed vertical saccadic slowing without oculomotor apraxia. Motor and sensory examination was normal; however, she manifested generalized areflexia. A nerve conduction study showed reduced sensory nerve action potential in the bilateral sural nerves with normal motor nerve conduction, which is compatible with pure sensory polyneuropathy. In the laboratory tests, the AFP level was elevated at 33.02 ng/mL (normal range 0.89–8.78 ng/mL). Cholesterol, albumin, and creatinine kinase levels were normal. Brain MRI demonstrated diffuse pontocerebellar atrophy (Figure 1A).
Early-onset progressive cerebellar ataxia with elevated AFP prompted us to perform a next-generation sequencing (NGS) ataxia gene panel composed of 39 genes, including genes related to AT, AOA1, AOA2, and AOA4. However, we only identified a heterozygous pathogenic splicing variant (c.6106+1G>A) in the SETX gene (NM_015046.7). Based on the high clinical suspicion of AOA2 and the known possibility of CNV in the pathogenicity of AOA2, we decided to search for additional genetic abnormalities related to AOA2. Whole-exome sequencing (WES) was performed, and it did not reveal any disease-causing variants in genes other than SETX. From the WES data, CNV analysis was performed using an in-house developed WES-based CNV detection tool. As a result, deletion of exon 26 in SETX was suspected in our patient. Confirmatory tests were performed by semiquantitative multiplex polymerase chain reaction. Segregation analysis indicated that the patient’s father carried the same splicing variant. However, neither parent had the exon 26 deletion in SETX, indicating a de novo mutation in our patient (Figure 1B). As a result, the patient was genetically confirmed to have AOA2.
AOA2 is rare in Korea, and to date, only one case has been reported [1]. The age of onset for AOA2 is between 3 and 30 years old, and the initial symptoms are mainly slowly progressing gait ataxia. Oculomotor apraxia occurs in only 50% of patients and is less frequent than in AOA1 (86%). Other oculomotor findings include increased horizontal saccade latencies, horizontal and vertical hypometria, and strabismus [2,3]. Movement symptoms such as head tremor, chorea, and dystonia may accompany these symptoms. Peripheral neuropathy mainly presents as axonal sensorimotor polyneuropathy, which occurs in more than 97% of patients with cerebellar atrophy. The AFP level was elevated in 98% of patients at first diagnosis, with a median serum level of 31 ng/mL. Serum cholesterol levels are elevated in 50% of cases, and serum creatinine kinase levels are elevated in patients with severe amyotrophy [2,4]. In our patient, gait disturbance started at the age of 15, and oculomotor apraxia was absent. Except for cerebellar dysfunctions, she had no other movement symptoms. This patient had pure sensory polyneuropathy.
The SETX gene encodes senataxin, which is a putative DNA/RNA helicase. Most of the mutations are missense and nonsense mutations, but large gene rearrangements have also been identified [4,5]. Mutations are often located in the helicase domain, and our patient’s mutation c.6106+1G>A in exon 15 was also in the helicase domain. This mutation affects RNA splicing and is likely to cause skipping of exon 15 [6]. Because exon 15 has 157 bp, the mutation will lead to an out-of-frame deletion. This deletion results in a premature stop codon and nonsense-mediated decay, which in turn prevents protein production. Because exon 26 is the last exon, the deletion of exon 26 in the opposite allele may not induce nonsense-mediated decay and is likely to generate a truncated protein (Figure 1C).
In previous studies, SETX gene analysis in AOA2 patients showed that most of the mutations are sequence variants. However, large mutations, including CNVs and deletions/insertions, are also reported in 8%–20% of patients, and these may escape routine diagnostics due to their size [5]. In our patient, AOA2 could not be confirmed by NGS gene panel alone. However, high clinical suspicion, including elevated AFP, led us to subsequent CNV analysis, and finally AOA2 was confirmed. Our case demonstrates that in circumstances where a clinical suspicion is high, additional CNV analysis, if available, should be considered in addition to conventional NGS. Moreover, if the CNV analysis is also negative, further analysis should be considered, such as searching for intronic variants through whole genome sequencing or evaluating SETX expression through RNA sequencing.
Notes
-
Ethics Statement
This study was approved by the Seoul National University Hospital Ethics Committee (IRB No. 2108-023-1241), and written informed consent was obtained from the patient.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding
This research was supported by a fund (2020-ER6904-01) of Korea Disease Control and Prevention Agency and the Diagnosis Support Programs for Rare Diseases funded by the Korea Disease Control and Prevention Agency.
-
Author Contributions
Conceptualization: Hee Jin Chang, Ryul Kim, Jangsup Moon, Man Jin Kim, Han-Joon Kim. Data curation: all authors. Formal analysis: Hee Jin Chang, Jangsup Moon, Man Jin Kim, Minchae Kim, Han-Joon Kim. Funding acquisition: Jangsup Moon, Man Jin Kim. Investigation: Hee Jin Chang, Jangsup Moon, Man Jin Kim, Han-Joon Kim. Methodology: Hee Jin Chang, Jangsup Moon, Man Jin Kim, Han-Joon Kim. Project administration: Han-Joon Kim. Supervision: Han-Joon Kim, Jangsup Moon, Man Jin Kim. Visualization: Jangsup Moon, Man Jin Kim, Minchae Kim. Writing—original draft: Hee Jin Chang. Writing—review & editing: all authors.
Figure 1.A: Diffuse pontocerebellar atrophy is demonstrated in T1-weighted brain MRI images. B: Segregation analysis indicates that the patient and father had the same variant, c.6160+1G>A, but in exon 26, the patient only showed a decreased normalized peak ratio. C: The location of mutations: c.6160+1G>A was in the helicase domain, and exon 26 was the last exon.
REFERENCES
- 1. Kim SH, Jang JH, Kim JS. Positional hemiseesaw nystagmus in ataxia with oculomotor apraxia type 2 due to a novel senataxin gene mutation: a new phenotype. Res Vestib Sci 2020;19:12–15.Article
- 2. Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 2009;132:2688–2698.ArticlePubMed
- 3. Jung I, Kim JS. Abnormal eye movements in parkinsonism and movement disorders. J Mov Disord 2019;12:1–13.ArticlePubMedPMC
- 4. Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Saccà F, Grieco GS, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 2006;66:1207–1210.ArticlePubMed
- 5. Bernard V, Minnerop M, Bürk K, Kreuz F, Gillessen-Kaesbach G, Zühlke C. Exon deletions and intragenic insertions are not rare in ataxia with oculomotor apraxia 2. BMC Med Genet 2009;10:87.ArticlePubMedPMC
- 6. Schöls L, Arning L, Schüle R, Epplen JT, Timmann D. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2). J Neurol 2008;255:495–501.ArticlePubMed
Citations
Citations to this article as recorded by

 E-submission
E-submission
 , Ryul Kim2
, Ryul Kim2
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
