E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 15(2); 2022 > Article
-
Case Report
Labrune’s Syndrome Presenting With Stereotypy-Like Movements and Psychosis: A Case Report and Review -
Chun-Yang Sim1
 , Shahizon Azura Mohamed Mukari2, Lock-Hock Ngu3, Chia-Yin Loh4, Rabani Remli5, Norlinah Mohamed Ibrahim5
, Shahizon Azura Mohamed Mukari2, Lock-Hock Ngu3, Chia-Yin Loh4, Rabani Remli5, Norlinah Mohamed Ibrahim5 -
Journal of Movement Disorders 2022;15(2):162-166.
DOI: https://doi.org/10.14802/jmd.21120
Published online: December 24, 2021
1Faculty of Medicine and Health Sciences, Universiti Malaysia Sarawak, Kota Samarahan, Malaysia
2Department of Radiology, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
3Department of Genetics, Kuala Lumpur Hospital, Kuala Lumpur, Malaysia
4CENTOGENE GmbH, Rostock, Germany
5Department of Medicine, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
- Corresponding author: Norlinah Mohamed Ibrahim, MRCP Department of Medicine, Universiti Kebangsaan Malaysia Medical Centre, Jalan Yaacob Latiff, Bandar Tun Razak, Kuala Lumpur 56000, Malaysia / Tel: +6012-2145306 / Fax: +603-91456640 / E-mail: norlinah@ppukm.ukm.edu.my
Copyright © 2022 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Labrune’s syndrome, or leukoencephalopathy with brain calcifications and cysts (LCC), is a rare genetic syndrome with variable neurological presentations. Psychiatric manifestations and involuntary movements are uncommonly reported. We report the case of a 19-year-old female, initially diagnosed with Fahr’s syndrome, who presented to us with acute psychosis, abnormal behavior and involuntary movements. Her brain computed tomography showed extensive bilateral intracranial calcifications without cysts. Genetic testing detected two compound heterozygous variants, NR_033294.1 n.*9C>T and n.24C>T, in the SNORD118 gene, confirming the diagnosis of LCC. We discuss the expanding phenotypic spectrum of LCC and provide a literature review on the current diagnosis and management of this rare syndrome.
- A 19-year-old female previously diagnosed with Fahr’s syndrome and epilepsy, first presented to Universiti Kebangsaan Malaysia Medical Centre in June 2018 with a three-day history of abnormal behavior associated with emotional lability, paranoia, irrelevant speech and a poor sleep pattern. There was gait instability and involuntary movements of her hands, mainly on the right. On examination, she was of short stature, had inappropriate affect, smiled and cried occasionally and did not obey commands. She had irrelevant speech with slurring of words. Neurological examination revealed no deficits or signs of meningism. There were choreoathetoid movements of her upper limbs (right > left), which appeared stereotyped and mimicked a traditional Malay dance movement (Supplementary Video 1 in the online-only Data Supplement). Brain computed tomography (CT) showed extensive bilateral calcifications. She refused lumbar puncture. An autoimmune screen for antinuclear antibody and anti-dsDNA were negative, and serum creatine kinase and lactate were normal. Initially, her serum Ca was raised, at 2.65 mmol/L, and her intact parathyroid hormone (iPTH) level was normal. When she was 18 years old, she had one episode of a low iPTH reading, at 1.43 pmol/L; however, subsequent serial iPTH and Ca levels were all within the normal range. Her PO4 level was normal, at 1.15 mmol/L.
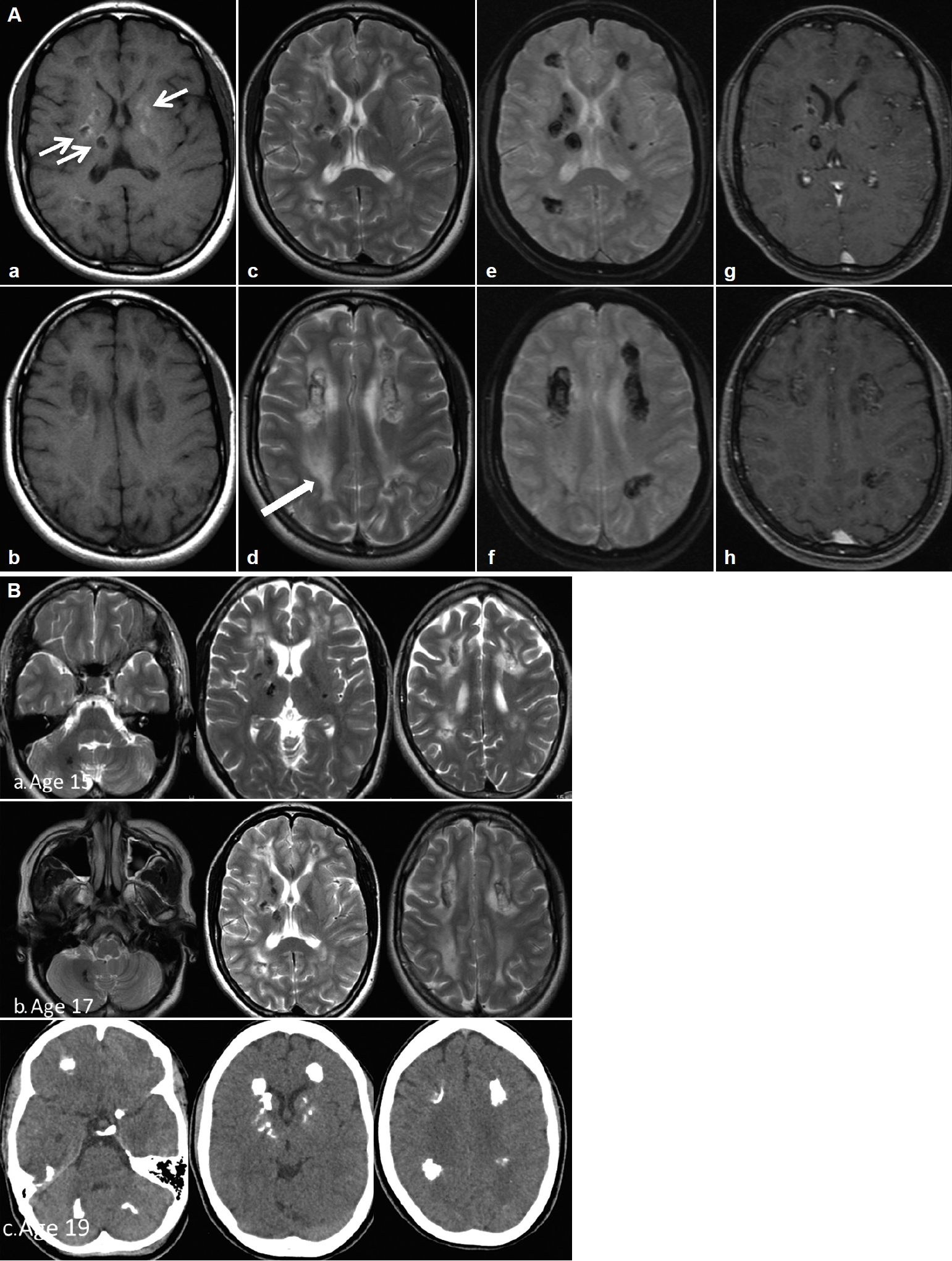
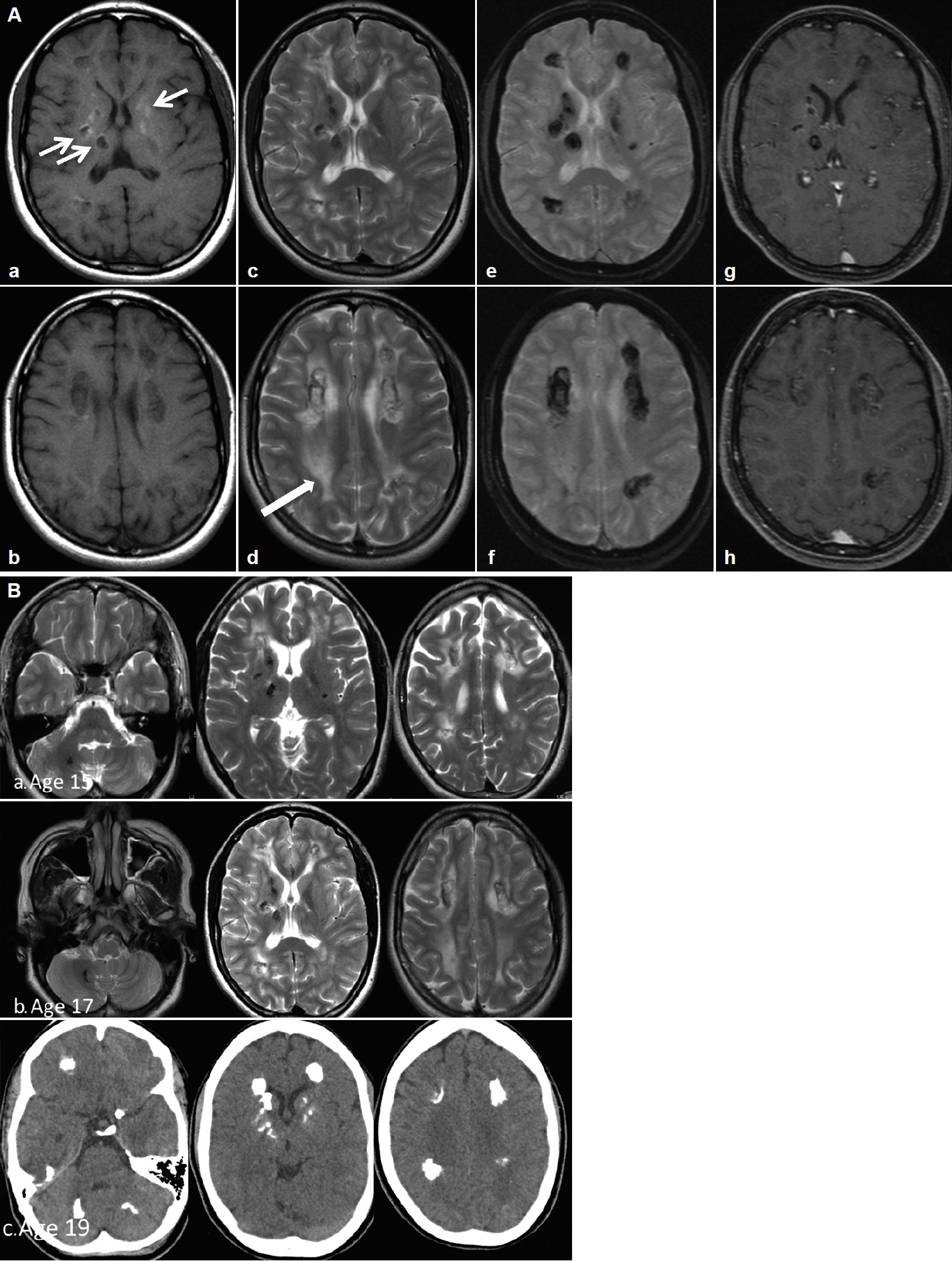
- Her perinatal history was unremarkable except for a single episode of seizure on the 25th day of life and a febrile seizure at 4 months. Her parents were nonconsanguineous. She had learning difficulties and was slow in reading and writing compared to her peers, although she was able to attend normal school until the age of 12. She has an older sister who is asymptomatic and a younger sister who is also a slow learner. She presented with a single episode of generalized tonic-clonic seizure at 15 years of age and later became more of focal aware seizures. Antiepileptic treatment was initiated, but she was noncompliant. A comprehensive neuropsychological assessment at age 17, using the Wechsler Intelligence Scale for Children Fourth Edition (WISCIV), revealed an IQ of 46, which fell in the extremely low range. At 18 years of age, she presented to another center with acute psychosis and behavioral changes associated with urinary and bowel incontinence and was noted to walk ‘stiffly and backward.’ She was treated for meningoencephalitis but did not complete treatment, as her family prematurely requested discharge to seek traditional remedies. She gradually returned to her normal self after a period of approximately 5–6 months and remained well until her current presentation. Serial brain imaging showed progressive, extensive bilateral intraparenchymal calcifications, mainly in the frontal and periventricular regions and the dentate nucleus. The calcifications in the deep white matter and the periventricular regions were associated with confluent asymmetrical white matter hyperintensities without a significant mass effect, representing leukoencephalopathies (Figure 1).
- An empirical diagnosis of a mitochondrial disorder was considered given her short stature, history of seizures and encephalopathic presentations associated with brain calcifications. A referral to a geneticist led to further genetic workup using wholegenome sequencing, which detected two recessively inherited compound heterozygous variants, NR_033294.1 n.*9C>T and n.24C>T, in the nonprotein coding gene SNORD118, diagnostic of LCC. Three weeks following her admission, her psychosis and stereotypy-like movements had resolved, and she was able to ambulate. During the latest clinical review 3 years later at the age of 22, she had returned to her baseline level, with no evidence of encephalopathy. She is currently learning Mandarin and Japanese languages and remains well, with infrequent seizures.
CASE REPORT
- LCC was first reported by Labrune et al. [1] in 1996. LCC is a rare microangiopathic disorder causing extensive calcifications and cyst formation in the brain. The genetic mutation was later identified in 2016 by Jenkinson et al. [2] as biallelic mutations in the SNORD118 gene, which leads to cerebral microangiopathy, widespread intracranial leukoencephalopathy, calcifications and cysts. LCC usually presents in the young, although the onset age varies widely from 1 month to 71 years [3]. Presently, 92 cases have been reported worldwide, with variable clinical presentations, including developmental delay, seizures, intracranial hypertension, headaches, cognitive decline, visual changes, speech disturbances, cerebellar signs, pyramidal and extrapyramidal symptoms (dystonia), and ischemic and hemorrhagic strokes [3]. Less common presentations include dysmorphism with intellectual disability [4,5]. Psychosis with altered behavior, including involuntary crying and laughing, as observed in our patient, was reported in only one other case [6]. Choreiform movements had not been reported previously. Our patient presented with recurrent episodes of psychosis and altered behavior, suggestive of encephalitis or encephalopathy, which resolved completely each time. These episodic events could be a sequela of postictal phenomena. Another explanation maybe due to the location of new lesions within the frontal lobe resulting in abnormal behavior, which gradually improved over time, mirroring the resolution of acute edema surrounding the new lesions.
- The SNORD118 gene, which is a nonprotein coding gene, encodes the box C/D snoRNA U8. U8 is suggested to be critical for the maturation of the 5.8S and 28S ribosomal RNAs (rRNAs) that comprise the large ribosomal subunit [5]. To date, several studies have reported the identification of recessively inherited biallelic SNORD118 mutations in LCC patients [2,5]. The worldwide prevalence of SNORD118 mutations is unknown. Cullinane et al. [7] reported an Irish traveler clan as possible carriers of the n.*5C>G mutation, while Iwama et al. [5] reported six unrelated Japanese families and controls with n.39G>C mutations. We detected two compound heterozygous variants in SNORD118 (n.*9C>T and n.24C>T) in our patient. The minor allele frequency for n.*9C>T and n.24C>T is 0.31% and 0.07%, respectively. Jenkinson et al. [2] reported the n.*9C>T substitution mutation in six patients with LCC of various nationalities, mostly European, including two British, one Belgian, one German, one French, and one North American individual. The second variant n.24C>T found in our patient was previously reported by Iwama et al. [5] in a patient of Filipino-Japanese ethnicity who also had an additional mutation in the n.3C>T allele. The patient presented with spastic hemiplegia, dystonia, speech disturbances and mild intellectual disability [5]. To date, there are no reports of patients with the same biallelic mutations in the SNORD118 gene as found in our patient. The wide variation in clinical manifestations of LCC is probably a reflection of the underlying structural damage associated with the location and distribution of the intracranial lesions rather than the genetic variants. Phenotypic variability in siblings carrying the same homozygous mutations was reported, indicating variable expressivity [7].
- The brain images of our patient did not demonstrate any cysts; hence, our patient did not fulfill the three cardinal radiological signs of this syndrome. However, two cases of Labrune’s syndrome without cysts were previously reported [4], and the authors proposed that remarkable leukoencephalopathy with brain calcifications, regardless of brain cysts or extra neurological features is pathognomonic for LCC [4]. It is postulated that cyst formation is a sequela of obliterative microangiopathy and necrotic processes that usually occur later in the disease course. Considering this radiological spectrum and the genetic mutations uncovered, LCC could be considered a form of hereditary cerebral small-vessel disease (HCSVD) similar to cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) or cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), which leads to microangiopathic leukoencephalopathies, although brain calcifications are not present in the latter two syndromes. Other genetic diseases that may cause brain calcifications include Coats plus syndrome, also known as cerebroretinal microangiopathy with calcifications and cysts (CRCC), and primary familial brain calcifications, or Fahr’s disease. CRCC is an autosomal-recessive syndrome that also leads to brain calcifications, leukoencephalopathy and brain parenchymal cysts, similar to LCC. Clinically, Coats plus syndrome is distinguished from LCC by the presence of exudative retinopathy and widespread multiorgan involvement, such as osteopenia, anemia and portal hypertension [8]. On the other hand, Fahr’s disease is an autosomal-dominant inherited condition with extensive basal ganglia calcifications, without leukoencephalopathy or brain parenchymal cysts. Adult-onset leukoencephalopathy associated with CSF1R mutation may also lead to a lesser degree of brain calcifications on imaging in addition to classic leukoencephalopathy, but this is inherited in an autosomal-dominant manner and presents at a later age. Fahr’s disease and CSF1R leukoencephalopathy do not lead to microangiopathy. The causes of HCSVD syndromes are listed in Table 1.
- There are no established diagnostic criteria for LCC. Clinicians need a high index of suspicion and rely on temporal clinical progression and serial brain imaging before arriving at the diagnosis, often leading to diagnostic delay. Furthermore, prior to the identification of the causative gene mutation, brain biopsy was advocated for tissue diagnosis [9]. Such approaches are far from ideal, and the nonspecific pathological findings may not facilitate diagnosis. The treatment of LCC remains symptomatic and includes neurosurgical interventions such as the removal of cyst(s) with or without a drain insertion to relieve the increased intracranial pressure associated with the brain lesions [3]. However, the intracranial lesions typically recur, and close monitoring with serial brain imaging and symptom review is required. Medical management is mainly symptomatic with antiepileptics and antipsychotics. As LCC is a microangiopathic disease and is postulated to be caused by a malfunction in the signaling pathway of vascular endothelial growth factors (VEGFs) [10], bevacizumab, an anti-VEGF monoclonal antibody, was shown to be effective in reducing cyst volume and white matter lesions as well as achieving clinical improvements in one study [10]. Nonetheless, this treatment is still considered investigational and requires further validation in larger studies.
- In conclusion, this is the first report of Labrune’s syndrome in Malaysia and highlights the importance of including Labrune’s syndrome in the diagnostic workup of patients without a family history presenting with neurological syndromes associated with brain calcifications and leukoencephalopathy. Labrune’s syndrome has a wide phenotypic spectrum, including recurrent encephalopathy, psychosis and involuntary movements, as in our case. Appropriate genetic testing is recommended if there is a high index of suspicion, and consideration should be given not only to the protein-coding regions but also to functionally important noncoding regions. Treatment remains symptomatic, although bevacizumab is potentially promising and may lead to the regression of lesions.
DISCUSSION
Supplementary Materials
Supplementary Video Legends
-
Ethics Statement
Research ethics committee of the Universiti Kebangsaan Malaysia Medical Centre approved to waiver of this manuscript. Written informed consent was obtained from the patient for the publication of this case report and any accompanying images and videos.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None
-
Author Contributions
Conceptualization: Norlinah Mohamed Ibrahim, Chun-Yang Sim. Supervision: Norlinah Mohamed Ibrahim. Visualization: Shahizon Azura Mohamed Mukari. Writing—original draft: Chun-Yang Sim. Writing—review & editing: all authors.
Notes
- 1. Labrune P, Lacroix C, Goutières F, de Laveaucoupet J, Chevalier P, Zerah M, et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy. Neurology 1996;46:1297–1301.ArticlePubMed
- 2. Jenkinson EM, Rodero MP, Kasher PR, Uggenti C, Oojageer A, Goosey LC, et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat Genet 2016;48:1185–1192.ArticlePubMedPMC
- 3. Kobets A, Oriko D, Groves M, Robinson S, Cohen A. Surgical considerations in Labrune syndrome. Childs Nerv Syst 2021;37:1765–1770.ArticlePubMed
- 4. Hermens M, van der Knaap MS, Kamsteeg EJ, Willemsen MA. A brother and sister with intellectual disability and characteristic neuroimaging findings. Eur J Paediatr Neurol 2018;22:866–869.ArticlePubMed
- 5. Iwama K, Mizuguchi T, Takanashi JI, Shibayama H, Shichiji M, Ito S, et al. Identification of novel SNORD118 mutations in seven patients with leukoencephalopathy with brain calcifications and cysts. Clin Genet 2017;92:180–187.ArticlePubMed
- 6. Liu X, Zheng X, Sui Q, Xu W, Zee CS. Leukoencephalopathy with cerebral calcifications and cysts: clinical and pathological features in two adults. Acta Neurol Belg 2016;116:47–52.ArticlePubMed
- 7. Cullinane PW, Lynch SA, Marnane M. Phenotypic variability in leukoencephalopathy with brain calcifications and cysts: case report of siblings from an Irish traveller family with a homozygous SNORD118 mutation. J Mol Neurosci 2020;70:1354–1356.ArticlePubMed
- 8. Polvi A, Linnankivi T, Kivelä T, Herva R, Keating JP, Mäkitie O, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet 2012;90:540–549.ArticlePubMedPMC
- 9. Wang Y, Cheng G, Dong C, Zhang J, Meng Y. Adult-onset leukoencephalopathy, brain calcifications and cysts: a case report. J Med Case Rep 2013;7:151.ArticlePubMedPMC
- 10. Fay AJ, King AA, Shimony JS, Crow YJ, Brunstrom-Hernandez JE. Treatment of leukoencephalopathy with calcifications and cysts with bevacizumab. Pediatr Neurol 2017;71:56–59.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Leukoencephalopathy, calcifications, and cysts: Labrune syndrome
Andrew Waack, Jordan Norris, Kathryn Becker, Alastair Hoyt, Jason Schroeder
Radiology Case Reports.2023; 18(2): 584. CrossRef - Leukoencephalopathy with calcifications and cysts: A case report with literature review
Jingya Li, Chun Li, Qing Zhang, Chao Qiu
Neurological Sciences.2023; 44(8): 2715. CrossRef - Adult-Onset Genetic Leukoencephalopathies With Movement Disorders
Mu-Hui Fu, Yung-Yee Chang
Journal of Movement Disorders.2023; 16(2): 115. CrossRef - Expanding the Natural History of SNORD118-Related Ribosomopathy: Hints from an Early-Diagnosed Patient with Leukoencephalopathy with Calcifications and Cysts and Overview of the Literature
Davide Politano, Guido Catalano, Elena Pezzotti, Costanza Varesio, Fabio Sirchia, Antonella Casella, Elisa Rognone, Anna Pichiecchio, Renato Borgatti, Simona Orcesi
Genes.2023; 14(9): 1817. CrossRef - Case report: Clinical and neuroradiological longitudinal follow-up in Leukoencephalopathy with Calcifications and Cysts during treatment with bevacizumab
Elena Scaffei, Bianca Buchignani, Rosa Pasquariello, Paola Cristofani, Raffaello Canapicchi, Laura Biagi, Flavio Giordano, Emanuela De Marco, Yanick J. Crow, Roberta Battini
Frontiers in Neurology.2023;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite