E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 15(1); 2022 > Article
-
Case Report
Parainfectious Anti-Glial Fibrillary Acidic Protein-Associated Meningoencephalitis -
Jae Young Joo1
 , Dallah Yoo2, Tae-Beom Ahn1,2
, Dallah Yoo2, Tae-Beom Ahn1,2 -
Journal of Movement Disorders 2022;15(1):66-70.
DOI: https://doi.org/10.14802/jmd.21115
Published online: November 25, 2021
1Department of Medicine, Graduate School, Kyung Hee University, Seoul, Korea
2Department of Neurology, Kyung Hee University Hospital, Seoul, Korea
- Corresponding author: Dallah Yoo, MD Department of Neurology, Kyung Hee University Hospital, 23 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Korea / Tel: +82-2-958-1794 / Fax: +82-2-958-8490 / E-mail: youdalla@gmail.com
Copyright © 2022 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Movement disorders associated with glial fibrillary acidic protein (GFAP) autoantibodies have rarely been reported as ataxia or tremors. A 32-year-old man with headache and fever, initially diagnosed with viral meningoencephalitis, showed gradual improvement with empirical treatment. Two weeks after the illness, he suddenly developed orofacial, tongue, and neck dyskinesia accompanied by oculomotor abnormalities, which developed into severe generalized choreoballism. Brain magnetic resonance imaging (fluid-attenuated inversion recovery) showed signal hyperintensities in the bilateral globus pallidus interna. The clinical picture suggested an acute inflammatory trigger of secondary autoimmune encephalitis. The autoimmune antibody test was positive for GFAP, with the strongest reactivity in the cerebrospinal fluid (CSF) before treatment and decreased reactivity in serial CSF examinations during immunotherapy. Dyskinesia gradually improved to the extent that it could be controlled with only oral medications. This patient presented with parainfectious GFAP meningoencephalitis with distinctive clinical features and imaging findings.
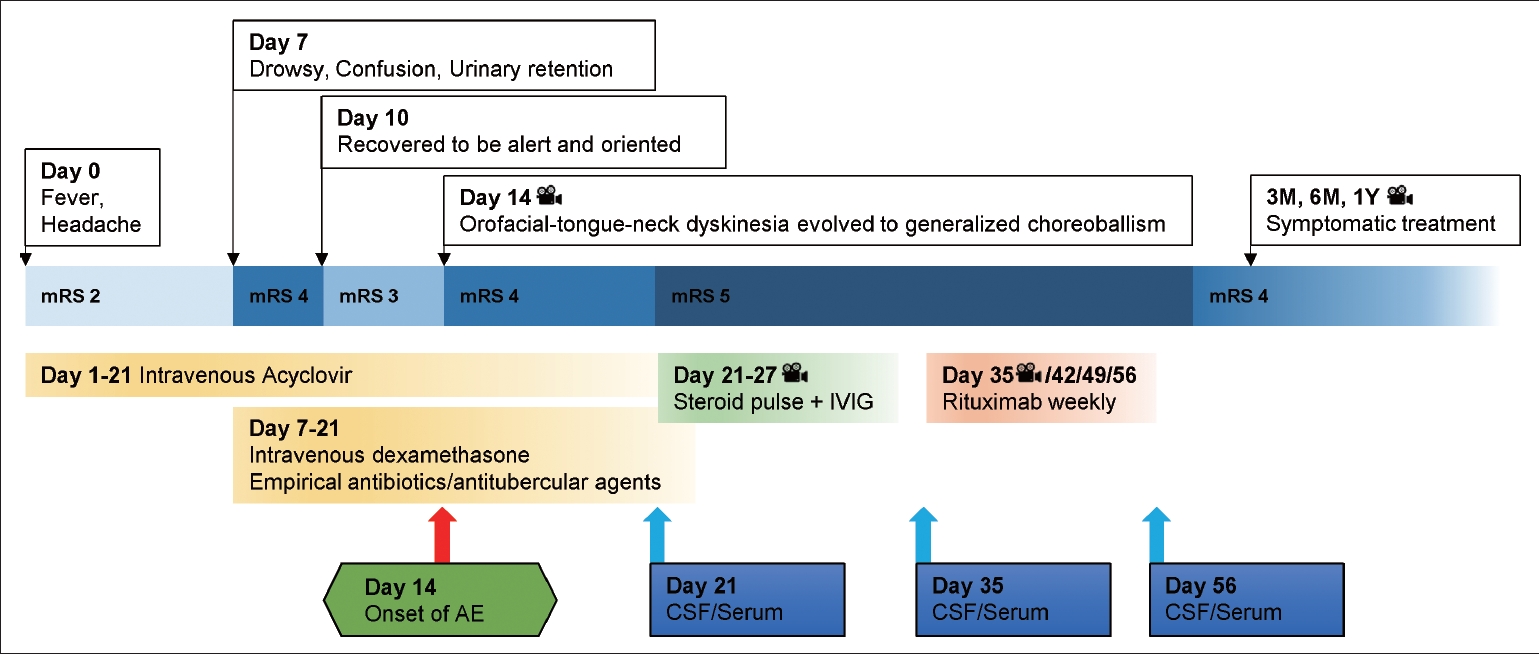
- A 32-year-old man presented to the emergency room with headache and fever (> 39°C) that started two days before admission (Day 0) (Figure 1). Neurological examinations showed neck stiffness and a positive Kernig’s sign. The CSF profiles indicated viral meningitis in which lymphocyte-dominant leukocytosis (white blood cell [WBC], 162 cells/μL; lymphocytes, 81%; neutrophils, 2%; monocytes, 17%), elevated protein concentration (111 mg/dL), and normal range of glucose (CSF 74 mg/dL, serum 105 mg/dL) were noted. Brain magnetic resonance imaging (MRI) revealed diffuse meningeal enhancement along the cerebral sulci without parenchymal involvement (Figure 2A). The patient was treated with intravenous acyclovir at a dose of 10 mg/kg three times a day. On day seven, his neurological status worsened, and drowsiness and confusion were noted, as was urinary retention. The patient temporarily increased his respiratory rate up to 40 breaths per minute, but the continuously monitored oxygen saturation remained above 95% as we immediately supplied oxygen with a facial mask and mechanical ventilator. Follow-up lumbar puncture revealed a WBC count of 648 cells/μL (lymphocytes, 71%; monocytes, 29%), a protein concentration of 258 mg/dL, and a glucose concentration of 47 mg/dL (serum glucose concentration, 155 mg/dL), consistent with clinical progression. Follow-up brain MRI showed no changes, and electroencephalography showed continuous slowing in the bilateral hemispheres. Treatment was initiated with 10 mg intravenous dexamethasone four times a day, empirical antibiotics, and antitubercular agents due to a high incidence of tuberculosis in South Korea. The following week, his neurological status gradually improved, and he was alert and oriented without motor or sensory deficits.
- On day 14, since the first onset of symptoms, he suddenly developed orofacial, tongue, and neck dyskinesia with abnormal vertical eye movements or reverse ocular dipping presenting as slow upward ocular drift and corrective fast downbeat nystagmus (Supplementary Video 1, segment 1 in the online-only Data Supplement). Dyskinesia evolved to continuous generalized choreoballism over the following week. He was alert and able to make eye contact and obey 1–2 step commands, although it was difficult for him to communicate in an intubated state. On day 21, severe dyskinesia was managed with a continuous infusion of midazolam and neuromuscular blockers such as rocuronium (Supplementary Video 1, segment 2 in the online-only Data Supplement). Follow-up CSF examination showed decreased inflammation (WBC, 90 cells/μL; lymphocytes, 87%, monocytes, 13%, protein concentration, 35 mg/dL, and glucose concentration, 68 mg/dL [serum glucose concentration, 106 mg/dL]). However, brain MRI showed subtle signal hyperintensity in the bilateral globus pallidus interna (GPi) on fluid-attenuated inversion recovery (FLAIR) without enhancement (Figure 2B). Although herpesvirus test results were negative in all three CSF examinations and other viral, bacterial, and tuberculosis etiologies were excluded (Supplementary Table 1 in the online-only Data Supplement), the clinical picture clearly suggested an inflammatory storm triggered by an acute infection in the central nervous system. The patient was treated with methylprednisolone (1 g per day for five days), intravenous immunoglobulin (0.4 g·kg-1·d-1 for five days), and rituximab (375 mg/mL weekly for a month). Follow-up CSF examinations on days 35 and 56 showed gradually decreasing leukocytosis and increasing protein levels (Supplementary Table 1 in the online-only Data Supplement). We referred autoimmune antibody tests to verified laboratories with CSF specimens from days 21, 35, and 56. The CSF was positive for the GFAP antibody using live cell-based assay techniques [4], with the sample from day 21 showing the strongest reactivity and the others showing gradually weaker reactivity. All types of neuronal surface antibodies were negative in all three samples.
- During immunomodulatory treatment, dyskinesia was maintained, although the doses of sedatives and neuromuscular blockers were slowly tapered off (Supplementary Video 1, segment 3 in the online-only Data Supplement). Tetrabenazine, haloperidol, clozapine, and diazepam were administered and showed benefits prior to immunotherapy, but the effects were not enough to stabilize his choreoballism. Topiramate, gabapentin, and pregabalin were briefly administered with initial mild benefits. Intravenous amantadine, tramadol, and valproate were ineffective. After rituximab treatment (four times weekly), follow-up brain MRI showed prominent signal hyperintensity at the bilateral GPi on FLAIR with signal hypointensity on T1-weighted images (Figure 2C). Diffuse cortical atrophies, including those of the mesial temporal lobes, were found in coronal sections of the brain images compared with images from the initial examination (Figure 2). A few months after the onset, involuntary movements of the face, neck, and limbs were gradually reduced through ongoing rehabilitation, but the patient remained functionally dependent because dyskinesia worsened during voluntary actions (Supplementary Video 1, segment 4 in the online-only Data Supplement). Articulation disability also improved, allowing some words to be understood; simple communication was possible for the patient. Regarding cognitive evaluation, he had an impairment in orientation and delayed word recall.
CASE REPORT
- The reported case is a parainfectious AE with CSF positive for GFAP antibody after infectious meningoencephalitis. Despite extensive diagnostic testing, the etiology of antecedent infection remains unknown. However, we diagnosed the patient with parainfectious AE triggered by an acute infection based on the following clinical features. First, a completely different neurological symptom developed while the clinical course improved. Second, leukocytosis gradually improved in the follow-up CSF tests. Third, the CSF was positive for the anti-GFAP antibody, with the strongest intensity before immunomodulatory treatment and weakening intensity over the course of and after immunotherapy. Finally, newly developed bilateral GPi lesions were highly correlated with the patient’s dyskinesia, and the abnormal movement was alleviated by second-line immunomodulatory treatment.
- Autoimmune GFAP astrocytopathy was first described in 2016 [5], and the frequent clinical phenotypes that were identified included subacute-onset meningeal symptoms (headache), encephalopathy (seizure, psychiatric symptoms, postural tremor, and ataxia), or myelopathic symptoms (numbness or weakness) [6]. Half of the patients showed striking radial linear perivascular enhancement on brain MRI, and patients with myelopathy showed a longitudinally extensive lesion on spine MRI. Neoplasms were detected in one-fourth of the patients, including ovarian teratoma, adenocarcinoma, and glioma. Autoimmune GFAP meningoencephalitis has rarely been reported in a patient after viral infection [7] or treatment with immune checkpoint inhibitors [8]. Most patients with GFAP antibodies in the CSF were responsive to immunotherapy, regardless of typical or atypical clinical features [5-8]. Although the present case had novel clinical and radiological phenotypes, responsiveness to immunotherapy was consistent with previous findings.
- Currently, the pathogenesis of GFAP astrocytopathy has not yet been clarified. In animal model studies, GFAP astrocytopathy is mediated by cytotoxic CD8+ T-cells specific for GFAP-derived peptides, and a viral trigger induces GFAP-specific CD8+ T-cells to exclusively target the meninges and vascular/perivascular space of the brain [9]. In humans, pathological studies have revealed extensive inflammatory changes with prominent perivascular infiltration by CD20+ B cells, CD8+ T-cells, and macrophages [10]. GFAP autoimmunity seems to support aberrant autoreactive T cell-mediated inflammation, contributing to the loss of astrocytes and neurons. However, the clinical presentations of anti-GFAP AE are heterogeneous, and the GFAP antibody presence has been described in various disorders, including traumatic brain injury, autism, and diabetes [6]. Other coexisting neural antibodies, including aquaporin-4 and N-methyl-D-aspartate receptor (NMDA-R) antibodies, are detected in one-fourth of patients with AE [5,6]. These findings indicate that GFAP autoimmunity occurs as a bystander or a secondary phenomenon with an unidentified mechanism.
- Movement disorders are associated with autoimmune GFAP astrocytopathy, including tremor, myoclonus, and ataxia [5,6]. To the best of our knowledge, this is the first report of generalized choreoballism in a patient with parainfectious, autoimmune GFAP astrocytopathy. In this case, severe dyskinesia resembled choreoathetosis seen in children with post-herpes simplex virus encephalitis NMDA-R AE [3]. Prominent bilateral GPi lesions are considered to be the underlying pathophysiology of hyperkinetic movements, in which the reduced inhibitory output from the damaged GPi could provoke disinhibition of undesired, competing motor patterns.
- Herein, we report a case of a patient with parainfectious AE confirmed to be GFAP-positive in the CSF; generalized dyskinesia improved with early immunotherapy. However, compared to reported features of anti-GFAP encephalitis, this case does not entirely correlate with the common clinical manifestations and therefore cannot rule out the possibility of antibodies of unknown significance. This study can broaden the clinical spectrum of autoimmune GFAP astrocytopathy, but further studies are needed to investigate GFAP antibody pathogenicity in AE.
DISCUSSION
Supplementary Materials
Video 1.
Supplementary Table 1.
-
Ethics Statement
All procedures performed in this study involving human participant were in accordance with the ethical standards of the institutional committee of Kyung Hee University Hospital and with the 1975 Helsinki declaration and its later amendments. Informed consent was obtained from the patient included in the study. The study was approved by the Institutional Review Board of Kyung Hee University Hospital (IRB No. 2021-02-012).
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None.
-
Author Contributions
Conceptualization: all authors. Data curation: Jae Young Joo, Dallah Yoo. Formal analysis: Jae Young Joo, Dallah Yoo. Investigation: Jae Young Joo, Dallah Yoo. Methodology: Jae Young Joo, Dallah Yoo. Supervision: Dallah Yoo, Tae-Beom Ahn. Validation: Dallah Yoo, Tae-Beom Ahn. Visualization: Jae Young Joo, Dallah Yoo. Writing—original draft: Jae Young Joo. Writing—review & editing: all authors.
Notes
- The authors thank the patient and his family members for their participation in this study. Furthermore, authors thank Dr. Josep Dalmau (the Institut d’Investigacions Biomèdiques August Pi i Sunyer [IDIBAPS]; Hospital Clínic, Department of Neurology; and Catalan Institute for Research and Advanced Studies [ICREA], Barcelona, Spain; Perelman School of Medicine, University of Pennsylvania, Philadelphia) for his generous support in antibody testing of glial fibrillary acidic protein (GFAP) and invaluable advice on patient care.
Acknowledgments


- 1. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840–851.ArticlePubMed
- 2. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638.ArticlePubMed
- 3. Armangue T, Spatola M, Vlagea A, Mattozzi S, Cárceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol 2018;17:760–772.PubMedPMC
- 4. Martinez-Hernandez E, Guasp M, García-Serra A, Maudes E, Ariño H, Sepulveda M, et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology 2020;94:e2302–e2310.ArticlePubMed
- 5. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol 2016;73:1297–1307.ArticlePubMed
- 6. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 2017;81:298–309.ArticlePubMed
- 7. Issa N, Martin C, Dulau C, Camou F. Severe anti-GFAP meningo-encephalomyelitis following viral infection. Mult Scler Relat Disord 2020;45:102448.ArticlePubMed
- 8. Kapadia RK, Ney DE, Hannan M, Farley M, Pastula DM, Piquet AL. Glial fibrillary acidic protein (GFAP) associated autoimmune meningoencephalitis in a patient receiving nivolumab. J Neuroimmunol 2020;344:577259.ArticlePubMed
- 9. Sasaki K, Bean A, Shah S, Schutten E, Huseby PG, Peters B, et al. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol 2014;192:3029–3042.ArticlePubMed
- 10. Long Y, Liang J, Xu H, Huang Q, Yang J, Gao C, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neurol 2018;25:477–483.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Relapsing Autoimmune GFAP Astrocytopathy: Case Report
Ekaterina O. Chekanova, Аlla А. Shabalina, Taras O. Simaniv, Rodion N. Konovalov, Larisa A. Dobrynina, Lyudmila A. Kalashnikova, Maria V. Gubanova, Maria N. Zakharova
Annals of Clinical and Experimental Neurology.2024; 17(4): 89. CrossRef - Comment on “Parainfectious Anti-Glial Fibrillary Acidic Protein-Associated Meningoencephalitis”
Byoung June Ahn, Kyum-Yil Kwon
Journal of Movement Disorders.2022; 15(2): 187. CrossRef - Re: Comment on “Parainfectious Anti-Glial Fibrillary Acidic Protein-Associated Meningoencephalitis”
Dallah Yoo, Tae-Beom Ahn
Journal of Movement Disorders.2022; 15(2): 189. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite