Department of Neurology, Bangur Institute of Neurosciences, Institute of Post Graduate Medical Education & Research (IPGME&R), Kolkata, India
Corresponding author: Atanu Biswas, MD, DM Department of Neurology, Bangur Institute of Neurosciences, Institute of Post Graduate Medical Education & Research (IPGME&R), 52/1A, S.N. Pandit Street, Kolkata 700025, India / Tel: +91-9836368139 / Fax: +91-33-2223-6677 / E-mail: atabis@gmail.com
• Received: October 14, 2020 • Revised: December 15, 2020 • Accepted: January 21, 2021
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Progressive myoclonic ataxia (PMA) is a rare disorder characterized by progressive ataxia and myoclonus, without significant cognitive decline and with or without infrequent seizures [1]. It is often difficult to distinguish PMA from progressive myoclonic epilepsy (PME), another rare entity characterized by a constellation of features including ataxia, myoclonus, cognitive decline, and seizures with or without other neurological deficits [2]. While ataxia and myoclonus are common features, the severity of seizures and cognitive decline appear to be the major differentiating points. Here, we describe a genetically proven sialidosis type 1 patient with overlapping features of the two syndromes. Additionally, our patient had psoriasis, which has never been reported before in sialidosis.
A 15-year-old boy born out of non-consanguineous parentage with normal birth and developmental history presented with tremors of both upper limbs for 1.5 years. Subsequently, he developed slurring of speech, unsteadiness of gait and swaying in all directions without limb weakness, sensory complaints, dizziness, hearing impairment or tinnitus. Within one year, the unsteadiness was further aggravated by sudden brief jerky movements of all four limbs that were precipitated by walking. He also had one episode of generalized tonic-clonic seizure (GTCS). There was no deterioration of his cognitive function. There was no history suggestive of connective tissue disorder, chronic diarrhea, recurrent chest infections or weight loss. There was no family history of similar complaints. Furthermore, he reported itchy lesions over his entire body over the past year.
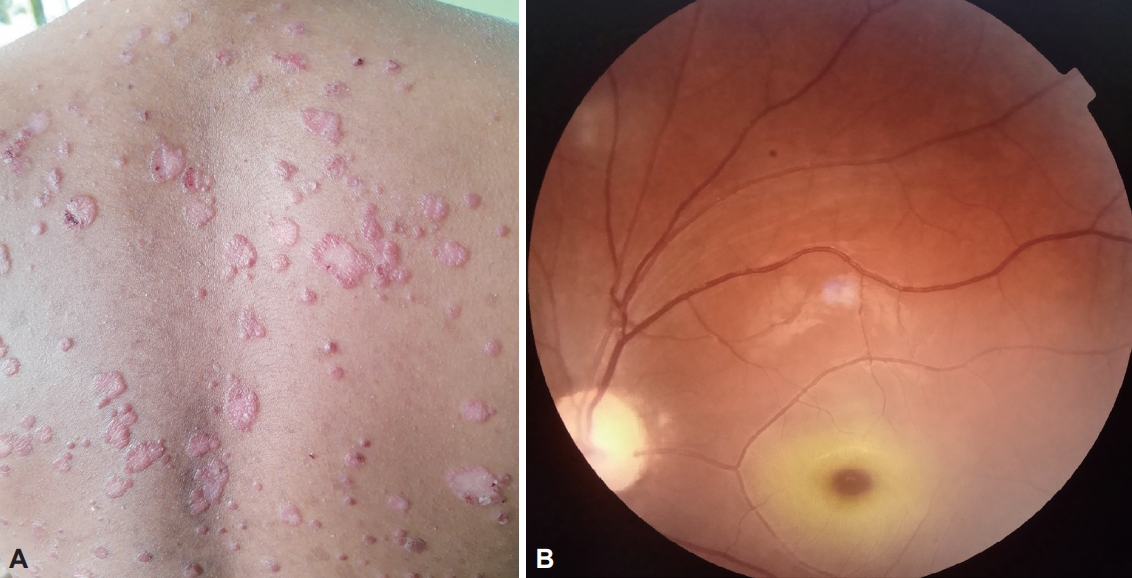
An examination revealed well-defined erythematous plaques with overlying micaceous scales on the trunk, extensor surface of the extremities, face and scalp with a positive Auspitz sign suggestive of psoriasis (Figure 1A). He scored 28/30 on the Mini-Mental State Examination (MMSE) and 96/100 on the Addenbrooke’s Cognitive Examination III (ACE-III). Neurological examination revealed dysarthria, severe gait ataxia and intention tremor in the upper limbs. Ocular examination revealed significant gaze-evoked nystagmus with bilateral smooth pursuit impairment and normal saccadic eye movements. Additionally, there were brief jerky movements of the upper and lower limbs at rest, which were mainly aggravated while walking, suggesting action myoclonus (Supplementary Video 1 in the online-only Data Supplement). Fundoscopy revealed bilateral macular cherry-red spots (Figure 1B).
Investigations including complete hemogram, blood glucose, thyroid profile, and renal and liver function tests were normal. Magnetic resonance imaging of the brain and cerebrospinal fluid tests were normal. The electroencephalogram showed short periodic generalized polyspike and wave epileptiform discharges independent of photostimulation. Visual evoked potential (VEP) studies showed bilateral prolonged P100 latencies, while somatosensory evoked potential study was normal. Wilson’s disease, mitochondrial, autoimmune and paraneoplastic conditions were ruled out. He had normal vitamin E and B12 levels but low serum zinc levels (49.82 μg/dL; range: 54–101 μg/dL). The light microscopic examination of a skin biopsy showed hyperkeratosis, follicular plugging, acanthosis and irregular downward elongation of rete ridges consistent with psoriasis. No inclusion bodies were detected in axillary skin biopsy. Nextgeneration sequencing revealed a homozygous missense variation in exon 5 of the neuraminidase 1 (NEU1) gene (chr6:g.318 27968T>C; c.872T>C), resulting in amino acid substitution of threonine for isoleucine at codon 291 (p. Ile291Thr; ENST000 00375631.4). This has been reported as a likely pathogenic variant consistent with sialidosis type 1, with in silico predictions being possibly damaging by PolyPhen-2 (HumDiv) and damaging by sorting intolerant from tolerant, likelihood ratio test and MutationTaster2. Enzyme assays could not be performed due to financial constraints.
He was managed with levetiracetam (20 mg/kg) and sodium valproate (30 mg/kg) with significant improvement in myoclonus (Supplementary Video 2 in the online-only Data Supplement) and no further GTCS. The skin lesions improved with zinc supplementation along with the application of local emollients, ketoconazole and steroid lotion.
The constellation of features including progressive and sequential development of cerebellar ataxia, action myoclonus and normal cognition with a single episode of GTCS in this boy suggested a clinical diagnosis of PMA. The presence of bilateral retino-optic pathway dysfunction in VEP and macular cherryred spots pointed towards a diagnosis of sialidosis, which is traditionally classified under PME. Detailed evaluation, including genetic studies, ultimately led to a diagnosis of sialidosis type 1, which presents with ataxia, myoclonus, visual impairment, tremors and cherry-red spots, as was seen in our case. It is a rare lysosomal storage disorder with autosomal recessive transmission, with a prevalence of <1 in 1,000,000, and is caused by a mutation in the NEU1 gene [3]. Deficiency of the NEU1 enzyme causes sialic acid-containing compounds to accumulate in lysosomes, affecting mainly the central nervous system, skeletal and reticuloendothelial systems.
Cognitive decline and frequent epilepsy were not seen in our patient, tilting the diagnosis in favor of PMA. In contrast, the presence of cherry-red spots in addition to ataxia and myoclonus favored PME. PMEs are known to progress into refractory epilepsy, while our patient had only a single episode of GTCS. While epileptologists prefer to lump these two conditions together, movement disorders experts want to split them. Protagonists lumping them together argue for these overlapping cases. However, as the etiological considerations under the two are different, many favor keeping them separate [1]. Adding to the nosological confusion, some authors have argued that certain disorders typically classified under PME, including Unverricht-Lundborg disease, mitochondrial encephalomyopathies, and sialidosis, among many other entities, are perhaps better categorized as PMAs where dementia and/or severe epilepsy is inconspicuous [4].
Additionally, our patient had psoriasis and zinc deficiency. Psoriasis has been reported only once in association with myoclonic epilepsy with ragged-red fibers [5]. Several authors have suggested the possible role of zinc deficiency in the pathogenesis of psoriasis and abnormal sphingolipid metabolism in causing inflammatory and dermatological diseases such as psoriasis, atopic dermatitis and icthyosis [6,7]. Whether the progressive accumulation of sialylated glycopeptides and oligosaccharides in sialidosis also contributes to psoriasis or its occurrence in this patient was merely an incidental finding needs further probing.
The complex presentation of a rare genetic disease, sialidosis type 1, makes our patient unique. The presence of psoriasis in the patient makes it even more interesting. The co-occurrence of these two conditions has not been reported in the literature, and a common pathogenetic mechanism remains unexplained.
Taken before the initiation of therapy, the video demonstrates a wide-based stance and gait with irregular cadence showing severe gait ataxia. Action myoclonus is evident in the form of jerky movements of the upper extremities and a bouncy gait. Intention tremor is evident during the finger-tonose test.
Video 2.
Taken during follow-up, the video shows a significant decrease in action myoclonus while walking. Subtle multifocal myoclonus is evident in the hands while at rest. Eye movement examination shows gaze-evoked nystagmus with bilateral smooth pursuit impairment.
Notes
Ethics statement
The authors confirm that written patient consent was obtained for this work in his own language. We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Conflicts of Interest
The authors have no financial conflicts of interest.
We acknowledge Dr. Arka Prava Chakraborty, Dr. Abhishek Chowdhury, and Dr. Arpan Dutta for their help in evaluating the case.
Figure 1.
A: Erythematous plaques with overlying micaceous scales on the back suggestive of psoriasis. B: Fundoscopy showing macular cherry-red spot.
REFERENCES
1. van der Veen S, Zutt R, Elting JWJ, Becker CE, de Koning TJ, Tijssen MAJ. Progressive myoclonus ataxia: time for a new definition? Mov Disord 2018;33:1281–1286.ArticlePubMedPMC
2. Satishchandra P, Sinha S. Progressive myoclonic epilepsy. Neurol India 2010;58:514–522.ArticlePubMed
4. Marsden CD, Obeso JA. Viewpoints on the Ramsay Hunt syndrome. The Ramsay Hunt syndrome is a useful clinical entity. Mov Disord 1989;4:6–12.ArticlePubMed
5. Finsterer J, Kovacs GG. Psoriasis, bulbar involvement, and diarrhea in late myoclonic epilepsy with ragged-red fibers-syndrome due to the m.8344A >G tRNA (Lys) mutation. Iran J Neurol 2017;16:45–49.PubMedPMC
6. Lei L, Su J, Chen J, Chen W, Chen X, Peng C. Abnormal serum copper and zinc levels in patients with psoriasis: a meta-analysis. Indian J Dermatol 2019;64:224–230.ArticlePubMedPMC
7. Moskot M, Bocheńska K, Jakóbkiewicz-Banecka J, Banecki B, Gabig-Cimińska M. Abnormal sphingolipid world in inflammation specific for lysosomal storage diseases and skin disorders. Int J Mol Sci 2018;19:247.ArticlePubMedPMC
Figure & Data
References
Citations
Citations to this article as recorded by
Novel Pathogenic Variant in the NEU1 Gene in a Patient With Sialidosis With Progressive Myoclonus Ataxia With Cherry-Red Spot Lulup K. Sahoo, Vidyasagar Kota, Pradeep K. Panigrahi, Srimant Pattnaik, Ajit P. Mishra, Srikanta K. Sahoo Neurology.2023; 101(19): 861. CrossRef
 E-submission
E-submission
 , Sougata Bhattacharya
, Sougata Bhattacharya
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
