E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 4(2); 2011 > Article
-
Case Report
Oromandibular Dyskinesia as the Initial Manifestation of Late-Onset Huntington Disease - Dong-Seok Oh, Eun-Seon Park, Seong-Min Choi, Byeong-Chae Kim, Myeong-Kyu Kim, Ki-Hyun Cho
-
Journal of Movement Disorders 2011;4(2):75-77.
DOI: https://doi.org/10.14802/jmd.11016
Published online: October 30, 2011
Department of Neurology, Chonnam National University Medical School, Gwangju, Korea
- Corresponding author: Seong-Min Choi, MD, Department of Neurology, Chonnam National University Hospital, 8 Hak-dong, Dong-gu, Gwangju 501-757, Korea, Tel +82-62-220-6171, Fax +82-62-228-3461, E-mail movement@chonnam.ac.kr
• Received: March 18, 2011 • Accepted: June 7, 2011
Copyright © 2011 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 62,708 Views
- 69 Download
- 4 Crossref
ABSTRACT
- Huntington’s disease (HD) is a neurodegenerative disorder characterized by a triad of choreoathetosis, dementia and dominant inheritance. The cause of HD is an expansion of CAG trinucleotide repeats in the HD gene. Typical age at onset of symptoms is in the 40s, but the disorder can manifest at any time. Late-onset (≥ 60 years) HD is clinically different from other adult or juvenile onset HD and characterized by mild motor problem as the initial symptoms, shorter disease duration, frequent lack of family history, and relatively low CAG repeats expansion. We report a case of an 80-year-old female with oromandibular dyskinesia as an initial manifestation of HD and 40 CAG repeats.
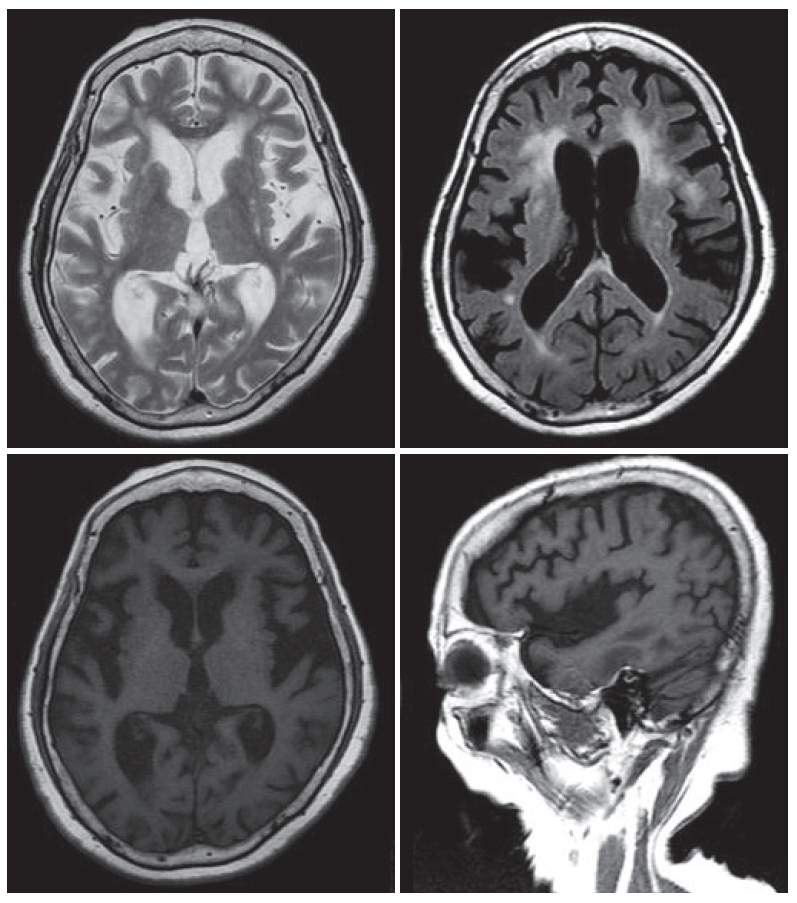
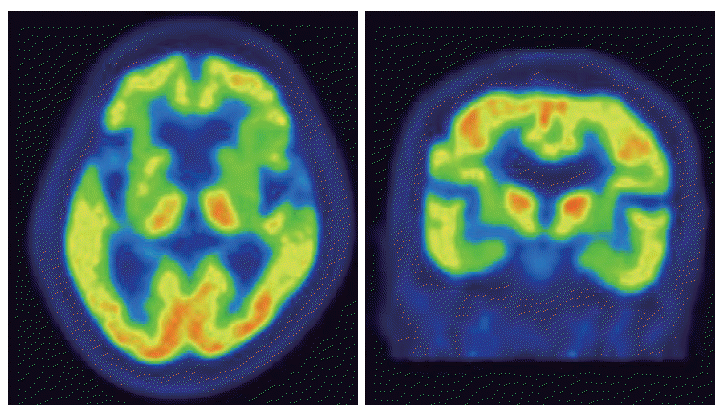
- An 80-year-old female visited our clinic due to involuntary movement on her oromandibular area and both extremities. She was diagnosed with hypertension and chronic small vessel disease about 10 years before and treated with clopidogrel 75 mg and amlodipine besylate 5 mg daily. Other family members including parents, siblings, 2 sons and 3 daughters were healthy and do not have any history of movement disorder. About 2 years before, she first experienced abnormal movement and discomfort on her oromandibular area. She ignored those symptoms as she had removed dental prosthesis 1 month before. However, those symptoms aggravated progressively. About 1 year later, abnormal involuntary movement that characterized by continuous restlessness was started on both extremities and frequent falling to forward was noted when she was walking. The restless movements worsened with stress and emotion, and were progressive, resulting in speech problems and gait disturbance. Caregivers also complained about memory decline and insomnia. On examination, she showed dysarthria and continuous OMD with generalized choreitic restlessness on her both extremities. Choreitic movement was spread to the trunk and affected sitting or standing posture. When she was ordered to maintain a sustained posture or to close her eyes, she could not maintain even longer than several seconds, which presenting motor impersistence. Cognitive function was also impaired, so it was very difficult to concentrate on or conduct doctor’s request. She scored 21 points on the Korean version of Mini-Mental State Examination. On detailed neuropsychological study (Seoul Neuropsychological Screening Battery), her cognitive function was impaired especially in comprehension, calculation, praxis, language and visuospatial memory and frontal lobe function. On laboratory evaluation including complete blood cell counts, routine chemistry, thyroid function, coagulation profile, tumor markers, peripheral blood smear, serum ceruloplasimin and 24 hour urine cupper were within normal range. Anti-nuclear antibody (ANA) was weak positive, but ANA titer has no clinical significance. Other auto immune studies were also normal. Brain magnetic resonance imaging (MRI) shows no specific findings except diffuse brain atrophy (Figure 1). Positron emission tomography using [18F]-fluoro-deoxyglucose shows severe hypometabolism in both basal ganglia (Figure 2). Genetic testing for HD revealed 40 CAG repeats on one huntington allele and 12 repeats on the other.
Case
- Here we report a patient with late-onset HD who presented OMD as an initial symptom. The typical age of onset for adult-onset HD is between the ages of 30 and 50,2 but the disorder can manifest at any time between infancy and senescence. The age of symptom onset is associated with CAG repeats length.3 There is negative correlation between the age of onset and the repeat length. In late-onset HD, the expansion size of CAG repeats is relatively low. In previous study, which reviewed thirty- four patients with late-onset HD (onset range 60–79 years), CAG trinucleotide expansion size ranged from 38–44 repeats. Another study noted that in persons beyond the age of 60, the effect of the CAG repeat length on age of onset seemed to diminish. 8 According to the study about the prevalence of late-onset HD, the expanded CAG repeat sequence was found with a narrow range of 36–38 repeats.6 It is also known that in lower CAG repeat reduced penetrance is present.7 Most of the patient were the first in their family to have a diagnosis of HD and, this case was also the first in her family.5 It is considered that the lack of family history of HD is related to the small CAG repeat size occurring in other family members.9,10
- Early signs of adult-onset HD are general restlessness, hygienic neglect, sleep disturbances, behavioral changes, anxiety, and depression.1 Motor signs follow and include involuntary movement that can be suppressed by the patient but not for long.1 In patients with late-onset HD, however, motor symptoms are most common first signs and presented with mild form.5 A study of late-onset HD also reported predominantly motor disturbance at onset with relatively mild disability and a favorable outlook for both independent living and for life expectancy. 6 Among the initial motor symptoms, there are typical facial movements with the characteristic raising of the eyebrows and the special facial expression of an astonished look.1 In this case, however, initial manifestation was OMD and progressed slowly to generalized chorea.
- OMD is abnormal, involuntary, aimless, repetitive movements affecting the tongue, lips, and jaw. It is often results from exposure to an offensive medication or certain orodental conditions. Misdiagnosis as a temporomandibular joint disorder or a psychogenic disease is not infrequent. The issue of the occurrence of ‘spontaneous’ (or unmedicated) OMD in normal aging remains blurred, since complete drug history and other dyskinesigenic conditions (e.g., orodental and cognitive status) are often incompletely documented.14 Other neurological disorders associated with OMD include chronic hepatic encephalopathy, infectious or paraneoplastic encephalitis, and subcortical infarcts. Rarely, OMD occurs in a variety of brain conditions, such as mental retardation, Rett syndrome, and neurodegenerative conditions like HD. So, it is not easy to diagnose HD when old patients present OMD as initial symptom. Typical finding in HD that contributes to overactivity is motor impersistence, the inability to maintain a voluntary muscle contraction at a constant level.11 Motor impersistence is independent of chorea and is linearly progressive, making it a possible surrogate marker of disease severity.12 It is a classic physical sign in HD that differentiates HD from other disorders, such as tardive dyskinesia.13 In this case, motor impersistence on tongue protrusion and on eye closing was noticed. So it is considerable to suspect late-onset HD when old patient presenting OMD and motor impersistence, even without other cognitive or psychiatric symptoms.
- We describe late onset HD patient with a 40 CAG repeats in the huntingtin gene and OMD as an initial manifestation. This case illustrates the difficulties in diagnosis of elderly patients with OMD. We suggest that HD should be considered in OMD patient with motor impersistence irrespective of the family history.
Discussion
Figure 1MRI of the patient. Brain MRI showed atrophic changes in the basal ganglia and cerebral cortex. Diffuse peri-ventricular white matter changes were also noted. MRI: magnetic resonance image.
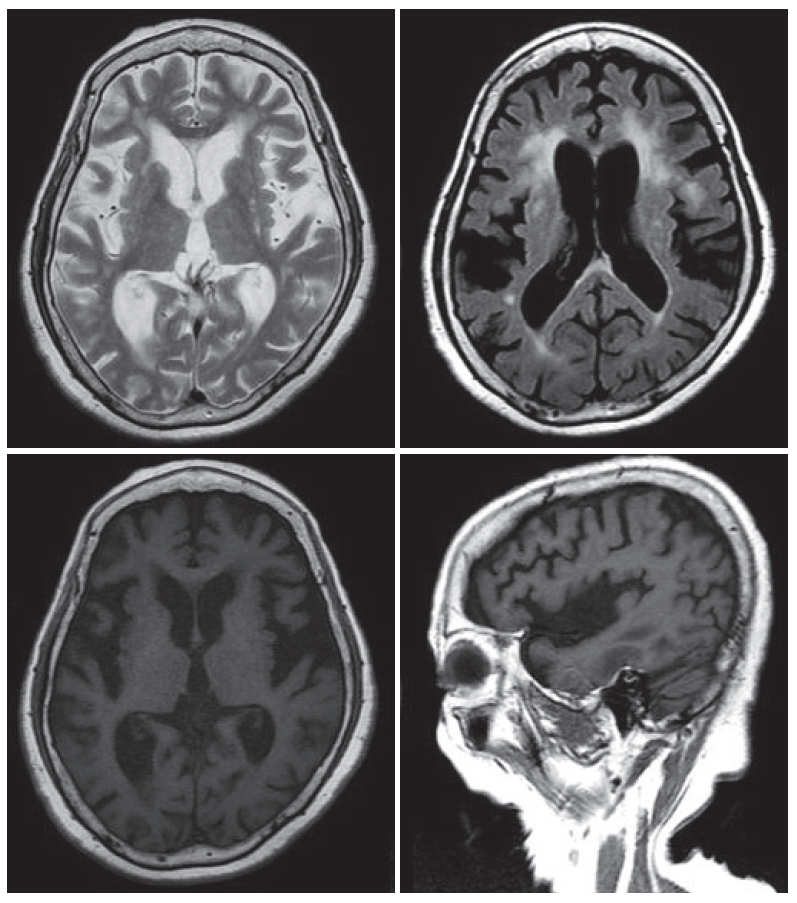
Figure 2[18F]-fluoro-deoxyglucose PET of the patients. Brain [18F]-fluoro-deoxyglucose PET showed severe hypometabolism in both basal ganglia. PET: positron emission tomography.

- 1. Joseph J, Eduardo T. Parkinson’s disease & movement disorders. In: Alexandra D, editor. Huntington’s disease. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007:236–245.
- 2. Kremer B. Clinical neurology of Huntington’s disease. In: Bates G, Harper P, Jones L, editors. Huntington’s Disease. 3rd ed. New York: Oxford University Press; 2002:30.
- 3. Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet 1993;4:398–403.ArticlePubMed
- 4. Groen JL, de Bie RM, Foncke EM, Roos RA, Leenders KL, Tijssen MA. Late-onset Huntington disease with intermediate CAG repeats: true or false? J Neurol Neurosurg Psychiatry 2010;81:228–230.ArticlePubMed
- 5. Lipe H, Bird T. Late onset Huntington Disease: clinical and genetic characteristics of 34 cases. J Neurol Sci 2009;276:159–162.ArticlePubMedPMC
- 6. James CM, Houlihan GD, Snell RG, Cheadle JP, Harper PS. Late-onset Huntington’s disease: a clinical and molecular study. Age Ageing 1994;23:445–448.ArticlePubMed
- 7. Walker FO. Huntington’s disease. Lancet 2007;369:218–228.ArticlePubMed
- 8. Kremer B, Squitieri F, Telenius H, Andrew SE, Theilmann J, Spence N, et al. Molecular analysis of late onset Huntington’s disease. J Med Genet 1993;30:991–995.ArticlePubMedPMC
- 9. Kartsaki E, Spanaki C, Tzagournissakis M, Petsakou A, Moschonas N, Macdonald M, et al. Late-onset and typical Huntington disease families from Crete have distinct genetic origins. Int J Mol Med 2006;17:335–346.ArticlePubMed
- 10. Bird TD, Lipe HP, Steinbart EJ. Geriatric neurogenetics: oxymoron or reality? Arch Neurol 2008;65:537–539.ArticlePubMedPMC
- 11. Gordon AM, Quinn L, Reilmann R, Marder K. Coordination of prehensile forces during precision grip in Huntington’s disease. Exp Neurol 2000;163:136–148.ArticlePubMed
- 12. Reilmann R, Kirsten F, Quinn L, Henningsen H, Marder K, Gordon AM. Objective assessment of progression in Huntington’s disease: a 3-year follow-up study. Neurology 2001;57:920–924.ArticlePubMed
- 13. Bhidayasiri R, Truong DD. Chorea and related disorders. Postgrad Med J 2004;80:527–534.ArticlePubMedPMC
- 14. Blanchet PJ, Rompré PH, Lavigne GJ, Lamarche C. Oral dyskinesia: a clinical overview. Int J Prosthodont 2005;18:10–19.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- The oral manifestations of Huntington's disease: A systematic review of prevalence
Luciana Munhoz, Ashjan Qasim Jabbar, William José e Silva Filho, Aline Yukari Nagai, Emiko Saito Arita
Oral Diseases.2023; 29(1): 62. CrossRef - Orofacial Dyskinesia and Intractable Hiccups in a Patient with Varicella-zoster Virus Encephalomyelitis
Akito Funatsu, Yohei Yamamoto, Midori Araki, Fumitoshi Aga, Hideki Mine
Internal Medicine.2023; 62(1): 119. CrossRef - Harmine prevents 3-nitropropionic acid-induced neurotoxicity in rats via enhancing NRF2-mediated signaling: Involvement of p21 and AMPK
Mohamed Z. Habib, Mariane G. Tadros, Hadwa A. Abd-Alkhalek, Magda I. Mohamad, Dalia M. Eid, Fatma E. Hassan, Hend Elhelaly, Yasser el Faramawy, Sawsan Aboul-Fotouh
European Journal of Pharmacology.2022; 927: 175046. CrossRef - Management of Traumatic Ulcerations of Lips in a Case of Huntington’s Disease: A Novel Application of Essix Retainer
Mohamed Iqbal J
Journal of Indian Orthodontic Society.2021; 55(4): 415. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite