E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 13(1); 2020 > Article
-
Review Article
Immunotherapy Targeting Neurodegenerative Proteinopathies: α-Synucleinopathies and Tauopathies -
Junghwan Shin
 , Han-Joon Kim, Beomseok Jeon
, Han-Joon Kim, Beomseok Jeon -
Journal of Movement Disorders 2020;13(1):11-19.
DOI: https://doi.org/10.14802/jmd.19057
Published online: December 19, 2019
Department of Neurology and Movement Disorder Center, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
- Corresponding author: Han-Joon Kim, MD, PhD Department of Neurology, Seoul National University College of Medicine, 103 Daehak-ro, Jongno-gu, Seoul 03080, Korea / Tel: +82-2-2072-2278 / Fax: +82-2-3672-7553 / E-mail: movement@snu.ac.kr
Copyright © 2020 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- α-Synuclein and tau deposition in the central nervous system is responsible for various parkinsonian syndromes, including Parkinson’s disease, multiple system atrophy, dementia with Lewy bodies, progressive supranuclear palsy and corticobasal degeneration. Emerging evidence has suggested that pathologic α-synuclein and tau are transmitted from cell to cell and further accelerate the aggregation of pathologic proteins in neighboring cells. Furthermore, extracellular pathologic proteins have also been reported to provoke inflammatory responses that lead to neurodegeneration. Therefore, immunotherapies targeting extracellular α-synuclein and tau have been proposed as potential disease-modifying strategies. In this review, we summarize completed phase I trials and ongoing phase II trials of immunotherapies against α-synuclein and tau and further discuss concerns and hurdles to overcome in the future.
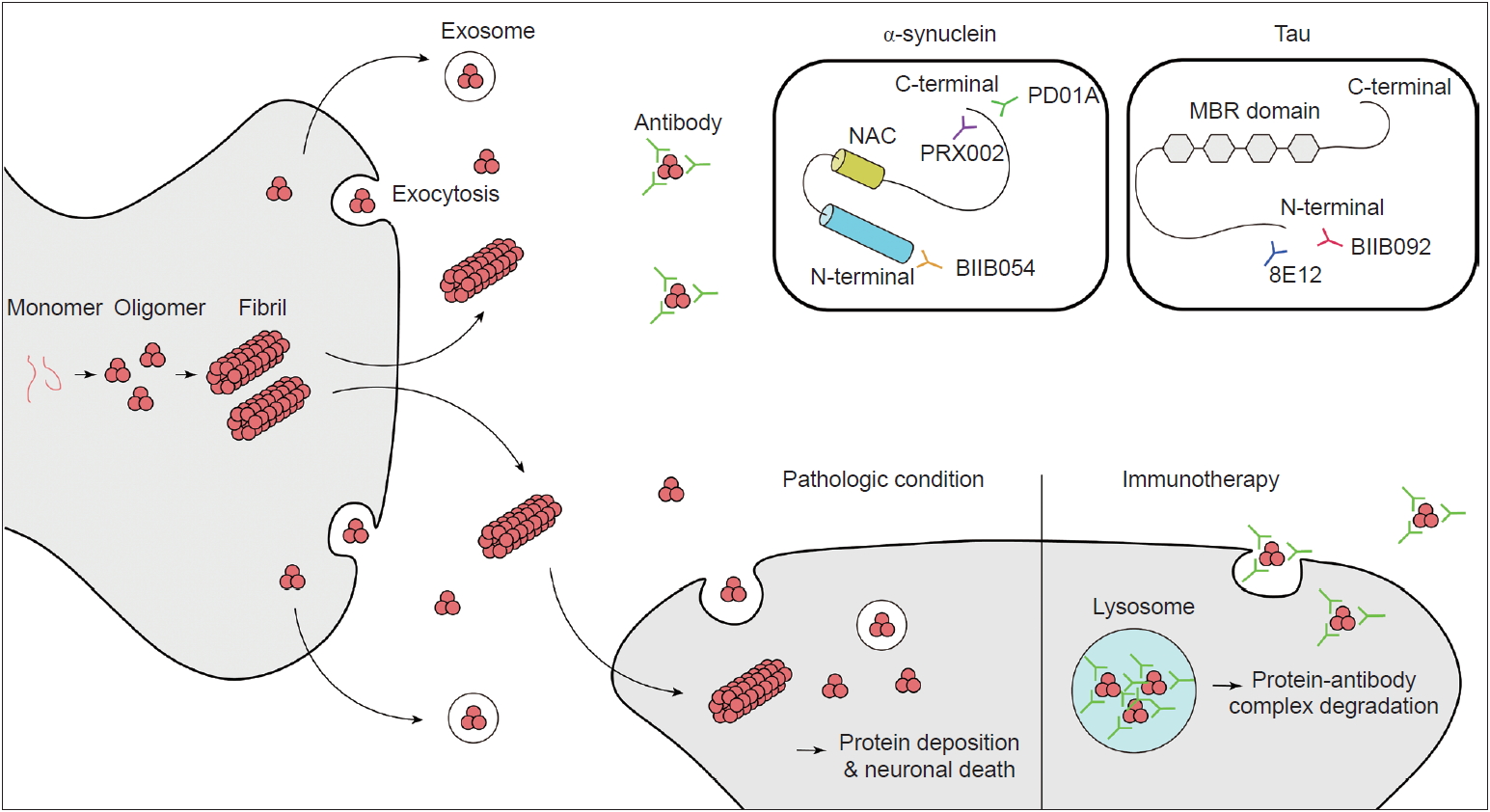
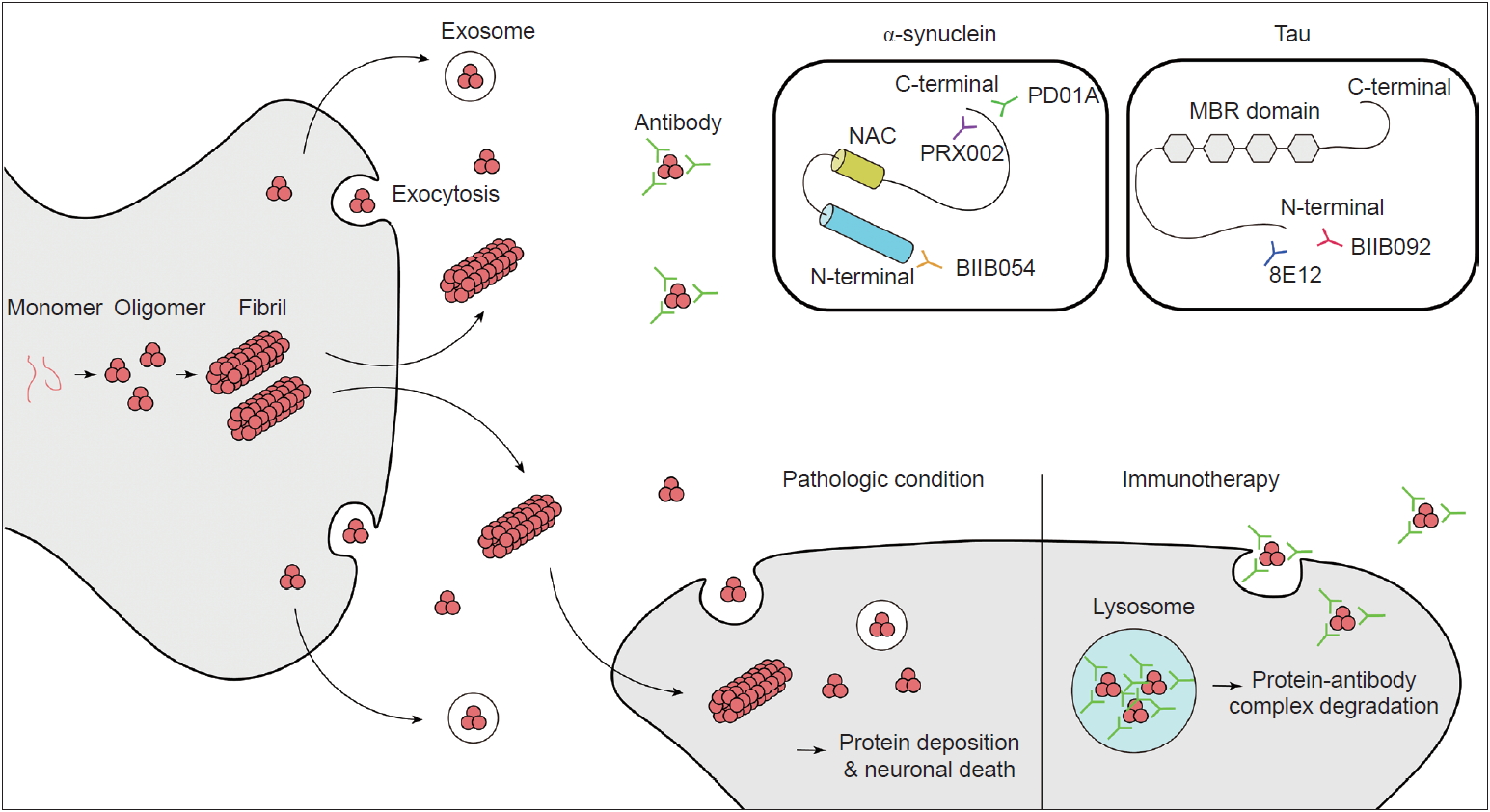
- α-Synuclein is a member of the synuclein family (which includes α, β, γ-synuclein and synoretin) and is translated from the SNCA gene located on chromosome 4q21-23 [7]. Synuclein was first discovered in cholinergic neurons in Torpedo californica (the Pacific electric ray) as a protein localized to synaptic vesicles and nuclei [24]. The name synuclein came from its localization to synapses and nuclei. α-Synuclein is highly expressed in the brain, especially in presynaptic terminals of neurons, as well as red blood cells and platelets [25,26]. Little is known about the physiologic role of native α-synuclein. However, studies have suggested that it is closely related to the regulation of synaptic vesicle dynamics [27]. It closely interacts with the SNARE (Soluble N-Ethylmaleimidesensitive factor Attachment protein Receptor) complex, which plays a critical role in the release of neurotransmitters [28,29]. Furthermore, there have been studies suggesting that α-synuclein plays a role in striatal dopamine release [30]. Its primary sequence is composed of 140 amino acids and contains 3 main domains: the N-terminal domain, C-terminal domain and non-amyloid-component (NAC) domain (Figure 1) [31]. The N-terminal domain is a highly conserved lysine-rich zone [32]. All previously reported mutations are in the N-terminal domain area, which shows that this region plays a critical role in aggregation. The NAC domain is a hydrophobic domain that enables α-synuclein to aggregate into β-sheet-rich fibrils. In the C-terminal region, the majority of posttranslational modifications, such as phosphorylation at serine 129, occurs [33,34], and truncation of this area is related to an increased rate of α- synuclein aggregation [35].
- There has been some debate about the structure of the native state of α-synuclein. It was previously thought to be unstructured in the native state; however, some studies have shown that it exists as a helically folded tetramer [36]. Under physiologically stressed conditions such as high temperature or low pH, α-synuclein adopts a partially folded intermediate structure [37]. As the partially folded structure contains hydrophobic patches on its surface, it is likely to be aggregated to form beta-sheet structures and form amyloid fibrils. As monomeric α-synuclein lacks hydrophobic patches, conditions that mediate the formation of the partially folded intermediate form are considered pathogenic [38]. The mechanism of the initial conformational change from normal monomeric α-synuclein to the pathologic form is largely unknown. However, it has been reported that fibril formation is accelerated in the presence of familial PD-associated α-synuclein mutations (E46K, A53T and H50Q), although some mutations (A30P, G51D and A53E) are associated with decreased fibril formation rates in vitro [39]. There are two phases of α-synuclein aggregation. One is primary nucleation, which is the formation of oligomers and fibrils from monomers; the other is the secondary nucleation phase, which is the elongation of fibrils or the transformation of oligomers from monomers by surface-catalyzed reaction by fibrils [39].
STRUCTURE AND FUNCTION OF α-SYNUCLEIN
- Other important questions are concern the form of α-synuclein that is toxic to neurons and the mechanism of this toxicity. Originally, it was thought that mature inclusion bodies cause neurodegeneration, but this has come under debate. Accumulating evidence has shown that it is the oligomeric intermediate that is toxic and causes neurodegeneration [40-42]. The main mechanism has been suggested to be increased membrane permeability, which leads to disruption of ionic homeostasis, depolarization of the mitochondrial membrane, and an increase in reactive oxygen species followed by caspase3 activation [43]. Additionally, the proposed mechanisms of α-synuclein toxicity include mitochondrial dysfunction [44-46], disruption of endoplasmic reticulum and Golgi trafficking [47,48], defective autophagy [49], disruption of interorganelle contacts [50] and altered transcription factor activation [51,52]. One of the amazing features of α-synuclein is that it can propagate from cell to cell (Figure 1). This was suggested by studies that demonstrated Lewy-body-like inclusions in grafted embryonic nigral neurons, which led to the hypothesis of the host-to-graft transmission of α-synuclein [53,54]. Similarly, in Braak’s autopsy study, it was hypothesized that α-synuclein aggregates spread to interconnected neural structures [10]. This observation cannot be readily explained if α-synuclein depositions occur spontaneously in each neuron. Subsequent studies have found that a portion of α-synuclein is present in vesicles in the cytoplasm and is secreted by exocytosis (Figure 1) [55]. Extracellular α-synuclein can exist as oligomers or fibrils, which can be internalized by neighboring neurons and undergo endosomal trafficking followed by lysosomal degradation [56,57]. If a fibril survives lysosomal degradation, it can accelerate α-synuclein aggregation through the mechanism mentioned above (secondary nuclearization). In addition, extracellular α-synuclein acts on the TLR2 signaling pathway in glial cells and produces an inflammatory response, which also plays a role in neurodegeneration [58]. Therefore, extracellular α-synuclein is currently considered a major pathogenic process of and a novel treatment target for synucleinopathies.
TOXICITY OF α-SYNUCLEIN AND ITS CELL-TO-CELL PROPAGATION
- Many antibodies have been studied and proposed as potential blockers of α-synuclein aggregation and propagation. The expected mechanisms are: 1) blocking extracellular α-synuclein in the brain to reduce cell-to-cell transmission, 2) reducing the total burden of the pathologic forms (oligomers/fibrils) of α-synuclein while sparing the physiologic forms (monomers/tetramers) of α-synuclein, and 3) stopping or slowing disease progression and hopefully reversing the symptoms of PD.
- There are two main ways of delivering antibodies against α-synuclein. One is passive immunization, and the other is active immunization. Several antibodies and vaccines (peptides used to induce an immune response to produce antibodies against α-synuclein) targeting the C-terminus or N-terminus of synuclein or synuclein oligomers have been studied in preclinical studies; these studies showed decreased neuronal loss with behavioral benefits in PD animal models [59-63]. Among them, several antibodies have been tested in human clinical trials. As of April 2019, phase I trials have been completed for two passive immunization antibodies and one active immunization peptide, and phase II trials are ongoing (Table 1 and 2).
- PRX002
- PRX002, also known as the 9E4 antibody, was the first therapeutic antibody developed by Prothena. The antibody targets the C-terminus of α-synuclein (Figure 1) and has more than 400-fold greater affinity for oligomeric α-synuclein than for monomeric α-synuclein. A preclinical animal model study showed that the passive delivery of the antibody blocked cell transmission, lowered intracellular α-synuclein pathology, and restored motor and cognitive deficits in multiple transgenic mouse models of PD [59,63]. Important findings were reported after the completion of the phase I trial [64]. The results showed that, at a dose of up to 60 mg/kg, it was tolerable and safe in PD patients. The serum levels of PRX002 were increased in a dose-dependent manner, and the serum α-synuclein level was reduced by up to 97% at the highest dose (60 mg/kg) [64]. The cerebrospinal fluid (CSF) level of PRX002 also increased proportionally with the dose. However, the ratio of the CSF PRX002 level and the serum PRX002 level was approximately 0.3% across all doses [64]. Interestingly, there was no change in the CSF α-synuclein level [64]. PRX002 is currently in a phase II clinical trial (PASADENA study, clinical trial identifier NCT03100149).
- BIIB054
- BIIB054 is a human anti-α-syn IgG1 monoclonal antibody also known as the 12F4 antibody. This antibody targets the N-terminus of α-synuclein (Figure 1). It is very selective for the aggregated form of α-synuclein (more than 800-fold) [65]. BIIB054 also showed good safety and tolerance at single doses up to 135 mg/ kg in healthy controls and 45 mg/kg in PD patients [65]. BIIB054 displayed dose-proportional pharmacokinetics and dose-dependently induced the formation of a BIIB054/α-synuclein complex in the serum [65]. The CSF-to-serum ratio of the antibody was between 0.3% (45 mg/kg group) and 0.5% (15 mg/kg group) when measured 4 weeks after injection in PD patients [65]. However, whether there was a significant decrease in the CSF α-synuclein level was not reported [65]. Currently, BIIB054 is being evaluated in a phase II study in early PD patients (SPARK study, clinical trial identifier NCT03318523).
- PD01A/PD03A
- Currently, there is only one company (AFFiRis, Wien, Austria) that has developed active immunization (a vaccine) targeting α-synuclein. α-Synuclein AFFITOPE (AFF) is a short peptide that mimics the α-synuclein molecule but has an amino acid sequence that is different from that of the native protein. It was designed to induce a B cell immunogenic response to produce an antibody without inducing a T cell-related autoimmune response [66]. PD01A and PD03A induced the robust production of antibodies that selectively recognize aggregated α-synuclein while sparing other synuclein family members and monomeric α-synuclein [66]. The peptide has been tested in animal models of synucleinopathies, including PD, MSA and DLB [66,67]. In animal models of synucleinopathies, vaccination reduced the burden of aggregated α-synuclein and neurodegeneration. Furthermore, it rescued the motor functions of PD and MSA model mice [66,67]. Most importantly, AFF did not induce any neuroinflammation or neural damage. AFF has undergone phase I clinical trials, including in both early PD and healthy controls (clinical trial identifier NCT01568099). Four injections of 15 µg or 75 µg of AFF peptide with aluminum oxyhydroxide adjuvant were administered every 4 weeks. The results showed a good safety profile without serious adverse events, and the participants were followed up for a longer period (52 weeks, clinical trial identifier NCT01885494). After successful phase I trials, participants underwent boost immunization with the peptide and showed clear and robust induction of α-synuclein antibodies after the first boost, and the second boost further stabilized the antibody titers [68]. PD01A-induced antibodies preferentially bind to both the oligomeric and fibril forms of α-synuclein rather than monomers. Furthermore, there was a trend for α-synuclein levels in the plasma and CSF to be reduced at week 26. However, the clinical scores of the vaccine-treated group did not show any difference compared with those of the untreated control group after 12 months of observation. Subsequent phase I studies have targeted both early PD (PD03) and early MSA patients (PD01/PD03).
IMMUNOTHERAPY INVOLVING α-SYNUCLEIN
- Tauopathies are a series of neurodegenerative disorders with various clinical and pathological presentations characterized by the aggregation of abnormal tau proteins. Tau plays a role in the polymerization and assembly of microtubules in the normal physiologic state [69]; therefore, tau is found in the axons of neurons and in glial cells, including astrocytes and oligodendrocytes [70]. There are 6 isoforms of tau, which are classified based on the number of microtubule binding-potential repeat domains (3R or 4R) and the number of amino-terminal inserts (0N, 1N or 2N) [71,72]. Phosphorylated or acetylated tau creates filamentous aggregates in neurons or glial cells, causing neurodegeneration. Tau normally exists in an equal ratio of 3R:4R tau in nondisease states, and tauopathies can be classified based on the ratio of 3R:4R tau [4]. Atypical parkinsonian syndromes, including PSP and CBD, are related to 4R dominant tauopathies [2,4]. Based on promising results from preclinical trials [73-75], anti-tau antibodies have been tested in clinical trials in patients with PSP or CBD. As of April 2019, there are three passive immunization tau antibodies (BIIB092, C2N-8E12 and UCB0107) for ongoing clinical trials (Table 2, 3). BIIB092 is a humanized IgG4 monoclonal anti-tau antibody developed by Biogen. A phase I trial of BIIB092 was completed in patients with PSP, and BIIB902 was reported to be safe in a multiple ascending dose study (clinical trial identifier NCT02460094). The antibody showed a dose-dependent accumulation in the serum and CSF [76]. There was a marked reduction in the CSF level of free eTau, which is an N-terminal tau fragment known to be related to the progression of the disease (Figure 1) [76]. The reduction rate was over 90% in all doses, and this reduction was maintained over 85 days [76]. BIIB092 is currently in a phase II trial aiming to recruit 396 patients with PSP (clinical trial identifier NCT03068468, PASSPORT). In addition, BIIB092 is currently in a phase Ib trial including CBD patients (clinical trial identifier NCT03658135, TauBasket). The 8E12 antibody is also a humanized IgG4 antibody developed by C2N Diagnostic and AbbVie. A phase I trial of the 8E12 antibody was completed (clinical trials identifier NCT02494024). The antibody has a half-life of 27 to 37 days, and a dose-dependent increase in the serum level was shown [77]. The CSF-to-serum ratio of the antibody was 0.18 to 0.35 [77]. The participants experienced no serious treatment-related adverse events [77]. Currently, the antibody is in phase II trials in PSP patients (clinical trials identifier NCT03391765).
IMMUNOTHERAPY AGAINST TAU
- There are some active clinical trials of therapies targeting α-synuclein and tau in parkinsonian syndromes, including PD, MSA, PSP and CBD. Based on all phase I trials of α-synuclein immunotherapies, antibodies and peptides show good tolerability without severe adverse events. Additionally, studies have consistently shown in a time- and dose-dependent manner. However, there may be some concerns. Phase I studies have shown a reduction in the serum α-synuclein concentration upon the formation of antibody/α-synuclein complexes. However, no study has shown a significant reduction in CSF α-synuclein levels. Furthermore, the ratio of the CSF concentration to the serum concentration of the antibody was less than 0.5% in passive immunization trials. The authors claimed that this was because a large portion of α-synuclein in the CSF is monomeric and that the PRX002 concentration in the CSF was not high enough to engage monomers, as PRX002 has a higher affinity (> 400-fold) for oligomers [64]. However, it remains unclear whether the CSF level of the antibody is sufficient with the currently used peripherally (intravenously) injected dose. The CSF penetration rate of other IgG monoclonal antibodies, including in preclinical animal studies, was reported to range from 0.1–1% [78-80]. Thus, determining optimal CSF antibody concentrations that can alter α-synuclein and tau propagation and deposition in humans would help establish an optimal peripheral (intravenous) dose of the antibody; this would require further studies regarding biomarkers of pathologic burden. On the other hand, we may have to consider alternative strategies, including direct intrathecal injection or increasing BBB penetration. Recently developed focused ultrasonography has been widely used to ablate the thalamus and STN in ET and PD patients [81,82]. One of the collateral damages is that it opens the local blood-brain barrier [83]. As this is a safe, noninvasive procedure, it might be considered an adjuvant strategy for peripheral passive immunization.
- The next concern relates to the efficacy of immunization. In AD, the amyloid beta-targeting antibodies bapineuzumab and solanezumab failed to stop cognitive decline after 72 weeks and 78 weeks, respectively [84,85]. A recently phase III trial of aducanumab, which is also an anti-amyloid beta antibody, was halted. Possible explanations for these results include that amyloid beta is an ineffective target compared with tau and that the participants in the trials exhibited disease that was too advanced for recovery. Considering the lessons learned from amyloid beta immunization trials in AD, there might be important points to consider.
- First, clinical trials should enroll patients in the early stage of disease. However, there are several hurdles to overcome. The diagnosis of PD, MSA and DLB is based on clinical evaluation, and there is a relatively high misdiagnosis rate (18–25%) [86-88]; he misdiagnosis rate tis even higher in patients in the early stage of the disease (35–47%) [89,90], who are suitable candidates for disease modifying treatment. There is a lack of biomarkers for early diagnosis and for monitoring the severity of the disease, which makes early diagnosis more difficult. Second, PD may manifest as a neurodegenerative disorder with mixed pathology that includes α-synuclein, tau and amyloid beta [91-93]. Therefore, clinically diagnosed and enrolled PD patients may be heterogeneous patients with different combinations of proteinopathies. Before considering immunotherapies targeting α-synuclein, it may be important to consider whether blocking and reducing the burden of α-synuclein alone is enough to ameliorate motor and nonmotor symptoms. Evaluating the correlation between clinical stage and the burden of α-synuclein aggregation, if possible, would lead to better targeted treatment strategies. In other words, without reliable biomarkers that reflect central α-synucleinopathy and the clinical stage, clinical trials of immunizations may repeatedly face similar limitations.
FUTURE DIRECTIONS
- Immunotherapies against α-synuclein and tau are promising disease-modifying treatment strategies with acceptable safety profiles. However, there are several hurdles to overcome for future clinical trials. These challenges include optimizing the delivery of antibodies to the brain, recruiting patients in the early stage of the disease and developing biomarkers that reflect central α-synuclein/ tau pathology.
CONCLUSION
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Junghwan Shin and Han-Joon Kim. Visualization: Junghwan Shin. Writing—original draft: Junghwan Shin. Writing—review & editing: All authors.
Notes
| Name | Type | Target of the antibody | Affinity (oligomeric/monomeric) | Trials | Dose | Half-life (d) | Change in serum α-syn level | Change in CSF α-syn level | BBB penetration rate | Safety |
|---|---|---|---|---|---|---|---|---|---|---|
| PRX002 | Passive | C-terminus | > 400 fold | Phase Ia | Single ascending dose | 18.2 | - | - | - | No severe or serious TEAEs |
| Phase Ib* | Multiple ascending doses | 10.2 | Up to 97% reduction | No | 0.3% | No severe or serious TEAEs | ||||
| BIIB054 | Passive | N-terminus | > 800 fold | Phase I† | Single ascending dose | 28–35 | - | - | 0.3–0.5% | No severe or serious TEAEs |
| PD01A | Active | C-terminus | - | Phase I | Single dose | - | - | - | - | No serious adverse events |
| Name | Type | Target of the antibody | Target disease | Trials | Dose | Half-life | Change in CSF tau level | BBB penetration rate | Safety |
|---|---|---|---|---|---|---|---|---|---|
| BIIB092 | Passive | N-terminus | Progressive supranuclear palsy | Phase Ia* | Single | 515–663 | > 80% reduction in eTau | - | No severe or serious TEAEs |
| Phase Ib | Multiple ascending doses | - | - | - | No severe or serious TEAEs | ||||
| 8E12 | Passive | N-terminus | Progressive supranuclear palsy | Phase I | Single ascending dose | 27–37 d | - | 0.18–0.35% | No severe or serious TEAEs |
* adapted from Qureshi et al. [76]
CSF: cerebrospinal fluid, BBB: blood-brain barrier, TEAEs: treatment-emergent adverse events.
- 1. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004;10 Suppl:S10–S17.ArticlePubMedPDF
- 2. Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 2009;8:1150–1157.ArticlePubMed
- 3. Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205–208.ArticlePubMed
- 4. Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 2011;45:384–389.ArticlePubMedPMCPDF
- 5. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol 2013;9:13–24.ArticlePubMedPDF
- 6. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840.ArticlePubMedPDF
- 7. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–2047.ArticlePubMed
- 8. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364:1167–1169.ArticlePubMed
- 9. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841.ArticlePubMed
- 10. Braak H, Del Tredici K. Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 2008;70:1916–1925.ArticlePubMed
- 11. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003;300:486–489.ArticlePubMed
- 12. Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012;149:1048–1059.ArticlePubMedPMC
- 13. Karpinar DP, Balija MB, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J 2009;28:3256–3268.ArticlePubMedPMC
- 14. Goedert M, Spillantini MG. Pathogenesis of the tauopathies. J Mol Neurosci 2011;45:425–431.ArticlePubMedPDF
- 15. Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol 2013;4:114.ArticlePubMedPMC
- 16. Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 2009;29:13578–13588.ArticlePubMedPMC
- 17. Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, et al. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013;136(Pt 7):2130–2146.ArticlePubMedPDF
- 18. Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 2013;110:E1817–E1826.ArticlePubMedPMC
- 19. Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, et al. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci 2011;31:6963–6971.ArticlePubMedPMC
- 20. Oueslati A, Schneider BL, Aebischer P, Lashuel HA. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc Natl Acad Sci U S A 2013;110:E3945–E3954.ArticlePubMedPMC
- 21. Lo Bianco C, Shorter J, Régulier E, Lashuel H, Iwatsubo T, Lindquist S, et al. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 2008;118:3087–3097.ArticlePubMedPMC
- 22. Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 2013;125:795–813.ArticlePubMedPMCPDF
- 23. Levin J, Schmidt F, Boehm C, Prix C, Bötzel K, Ryazanov S, et al. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol 2014;127:779–780.ArticlePubMedPMCPDF
- 24. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988;8:2804–2815.ArticlePubMedPMC
- 25. Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH, et al. Expression patterns of α-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells 2000;10:65–70.ArticlePubMedPDF
- 26. Ltic S, Perovic M, Mladenovic A, Raicevic N, Ruzdijic S, Rakic L, et al. Alpha-synuclein is expressed in different tissues during human fetal development. J Mol Neurosci 2004;22:199–204.ArticlePubMed
- 27. Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 2002;22:8797–8807.ArticlePubMedPMC
- 28. Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005;123:383–396.ArticlePubMed
- 29. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010;329:1663–1667.ArticlePubMedPMC
- 30. Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239–252.ArticlePubMed
- 31. Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 2015;14:855–866.ArticlePubMedPMC
- 32. Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 2011;108:17797–17802.ArticlePubMedPMC
- 33. Kellie JF, Higgs RE, Ryder JW, Major A, Beach TG, Adler CH, et al. Quantitative measurement of intact alpha-synuclein proteoforms from postmortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep 2014;4:5797.ArticlePubMedPMCPDF
- 34. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002;4:160–164.ArticlePubMedPDF
- 35. Hoyer W, Cherny D, Subramaniam V, Jovin TM. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 2004;43:16233–16242.ArticlePubMed
- 36. Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011;477:107–110.ArticlePubMedPMCPDF
- 37. Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 2001;276:10737–10744.ArticlePubMed
- 38. Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn 2003;21:211–234.ArticlePubMed
- 39. Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK. α-synuclein aggregation and its modulation. Int J Biol Macromol 2017;100:37–54.ArticlePubMed
- 40. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011;108:4194–4199.ArticlePubMedPMC
- 41. Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 2000;97:571–576.ArticlePubMedPMC
- 42. Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008;131:642–650.ArticlePubMedPDF
- 43. Shen J, Du T, Wang X, Duan C, Gao G, Zhang J, et al. α-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res 2014;1591:14–26.ArticlePubMed
- 44. Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 2010;29:3571–3589.ArticlePubMedPMC
- 45. Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 2011;286:20710–20726.ArticlePubMedPMC
- 46. Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 2014;289:21490–21507.ArticlePubMedPMC
- 47. Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003;302:1772–1775.ArticlePubMedPMC
- 48. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 2017;23:1–13.ArticlePMCPDF
- 49. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004;305:1292–1295.ArticlePubMed
- 50. Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, et al. α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 2014;34:249–259.ArticlePubMedPMC
- 51. Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010;2:52ra73.ArticlePubMedPMC
- 52. Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013;155:1351–1364.ArticlePubMedPMC
- 53. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy bodylike pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 2008;14:504–506.ArticlePubMedPDF
- 54. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008;14:501–503.ArticlePubMedPDF
- 55. Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein--a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 2014;10:92–98.ArticlePubMedPDF
- 56. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009;106:13010–13015.ArticlePubMedPMC
- 57. Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, et al. αSynuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 2011;121:715–725.ArticlePubMedPMC
- 58. Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013;4:1562.ArticlePubMedPMCPDF
- 59. Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 2011;6:e19338.ArticlePubMedPMC
- 60. Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005;46:857–868.ArticlePubMed
- 61. Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, et al. Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 2017;5:7.ArticlePubMedPMCPDF
- 62. Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, et al. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci 2012;32:13454–13469.ArticlePubMedPMC
- 63. Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 2014;34:9441–9454.ArticlePubMedPMC
- 64. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-Synuclein monoclonal antibody, in patients with parkinson disease: a randomized clinical trial. JAMA Neurol 2018;75:1206–1214.ArticlePubMedPMC
- 65. Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Mov Disord 2019;34:1154–1163.ArticlePubMedPMC
- 66. Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol 2014;127:861–879.ArticlePubMedPMCPDF
- 67. Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener 2015;10:10.ArticlePubMedPMCPDF
- 68. Zella SM, Metzdorf J, Ciftci E, Ostendorf F, Muhlack S, Gold R, et al. Emerging immunotherapies for Parkinson disease. Neurol Ther 2019;8:29–44.ArticlePubMedPDF
- 69. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 1975;72:1858–1862.ArticlePubMedPMC
- 70. Kahlson MA, Colodner KJ. Glial tau pathology in tauopathies: functional consequences. J Exp Neurosci 2015;9(S2):43–50.ArticlePMC
- 71. Andreadis A, Broderick JA, Kosik KS. Relative exon affinities and suboptimal splice site signals lead to non-equivalence of two cassette exons. Nucleic Acids Res 1995;23:3585–3593.ArticlePubMedPMCPDF
- 72. Goedert M, Spillantini MG, Davies SW. Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol 1998;8:619–632.ArticlePubMed
- 73. West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, et al. Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 2017;4:236–241.PubMed
- 74. Yanamandra K, Patel TK, Jiang H, Schindler S, Ulrich JD, Boxer AL, et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med 2017;9:pii: eaal2029.Article
- 75. Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013;80:402–414.ArticlePubMedPMC
- 76. Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y) 2018;4:746–755.ArticlePubMedPMC
- 77. Budur K, West T, Braunstein JB, Fogelman I, Bordelon YM, Litvan I, et al. Results of a Phase 1, single ascending dose, Placebo-controlled study of ABBV-8E12 in patients with progressive supranuclear palsy and Phase 2 study design in early Alzheimer’s disease. Alzheimers Dement 2017;13(Suppl):599–600.Article
- 78. Pepinsky RB, Shao Z, Ji B, Wang Q, Meng G, Walus L, et al. Exposure levels of anti-LINGO-1 Li81 antibody in the central nervous system and dose-efficacy relationships in rat spinal cord remyelination models after systemic administration. J Pharmacol Exp Ther 2011;339:519–529.ArticlePubMed
- 79. Beigel JH, Nordstrom JL, Pillemer SR, Roncal C, Goldwater DR, Li H, et al. Safety and pharmacokinetics of single intravenous dose of MGAWN1, a novel monoclonal antibody to West Nile virus. Antimicrob Agents Chemother 2010;54:2431–2436.ArticlePubMedPMC
- 80. Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin Drug Metab Toxicol 2012;8:141–160.ArticlePubMed
- 81. Elias WJ, Lipsman N, Ondo WG, Ghanouni P, Kim YG, Lee W, et al. A randomized trial of focused ultrasound thalamotomy for essential tremor. N Engl J Med 2016;375:730–739.ArticlePubMed
- 82. Martinez-Fernandez R, Rodríguez-Rojas R, Del Álamo M, HernándezFernández F, Pineda-Pardo JA, Dileone M, et al. Focused ultrasound subthalamotomy in patients with asymmetric Parkinson’s disease: a pilot study. Lancet Neurol 2018;17:54–63.ArticlePubMed
- 83. Chu PC, Chai WY, Tsai CH, Kang ST, Yeh CK, Liu HL. Focused ultrasound-induced blood-brain barrier opening: association with mechanical index and cavitation index analyzed by dynamic contrast-enhanced magnetic-resonance imaging. Sci Rep 2016;6:33264.ArticlePubMedPMCPDF
- 84. Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322–333.ArticlePubMedPMC
- 85. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:311–321.ArticlePubMed
- 86. Litvan I, MacIntyre A, Goetz CG, Wenning GK, Jellinger K, Verny M, et al. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol 1998;55:969–978.ArticlePubMed
- 87. Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson’s disease. Lancet Neurol 2006;5:75–86.ArticlePubMed
- 88. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184.ArticlePubMedPMC
- 89. Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in parkinsonism--a prospective study. Can J Neurol Sci 1991;18:275–278.ArticlePubMed
- 90. Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014;83:406–412.ArticlePubMedPMC
- 91. Rochester L, Galna B, Lord S, Yarnall AJ, Morris R, Duncan G, et al. Decrease in Aβ42 predicts dopa-resistant gait progression in early Parkinson disease. Neurology 2017;88:1501–1511.ArticlePubMedPMC
- 92. Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003;300:636–640.ArticlePubMed
- 93. Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 2010;30:7281–7289.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Overlaps and divergences between tauopathies and synucleinopathies: a duet of neurodegeneration
Wen Li, Jia-Yi Li
Translational Neurodegeneration.2024;[Epub] CrossRef - Modeling the dynamics of innate and adaptive immune response to Parkinson's disease with immunotherapy
Salma M. Al-Tuwairqi, Asma A. Badrah
AIMS Mathematics.2023; 8(1): 1800. CrossRef - Evaluation of an Adoptive Cellular Therapy-Based Vaccine in a Transgenic Mouse Model of α-synucleinopathy
Winston T. Chu, Jesse Hall, Anjela Gurrala, Alexander Becsey, Shreya Raman, Michael S. Okun, Catherine T. Flores, Benoit I. Giasson, David E. Vaillancourt, Vinata Vedam-Mai
ACS Chemical Neuroscience.2023; 14(2): 235. CrossRef - Direct digital sensing of protein biomarkers in solution
Georg Krainer, Kadi L. Saar, William E. Arter, Timothy J. Welsh, Magdalena A. Czekalska, Raphaël P. B. Jacquat, Quentin Peter, Walther C. Traberg, Arvind Pujari, Akhila K. Jayaram, Pavankumar Challa, Christopher G. Taylor, Lize-Mari van der Linden, Titus
Nature Communications.2023;[Epub] CrossRef - Inflammation in multiple system atrophy
Marta Leńska-Mieciek, Natalia Madetko-Alster, Piotr Alster, Leszek Królicki, Urszula Fiszer, Dariusz Koziorowski
Frontiers in Immunology.2023;[Epub] CrossRef - Immunisation with UB-312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α-synuclein accumulation in the brain and gut
Jacqui T. Nimmo, Harry Smith, Chang Yi Wang, Jessica L. Teeling, James A. R. Nicoll, Ajay Verma, Jean-Cosme Dodart, Zhi Liu, Feng Lin, Roxana O. Carare
Acta Neuropathologica.2022; 143(1): 55. CrossRef - Efficacy and immunogenicity of MultiTEP-based DNA vaccines targeting human α-synuclein: prelude for IND enabling studies
Changyoun Kim, Armine Hovakimyan, Karen Zagorski, Tatevik Antonyan, Irina Petrushina, Hayk Davtyan, Gor Chailyan, Jonathan Hasselmann, Michiyo Iba, Anthony Adame, Edward Rockenstein, Marcell Szabo, Mathew Blurton-Jones, David H. Cribbs, Anahit Ghochikyan,
npj Vaccines.2022;[Epub] CrossRef - Evidence of Inflammation in Parkinson’s Disease and Its Contribution to Synucleinopathy
Thuy Thi Lai, Yun Joong Kim, Hyeo-il Ma, Young Eun Kim
Journal of Movement Disorders.2022; 15(1): 1. CrossRef - Slowing Parkinson’s Disease Progression with Vaccination and Other Immunotherapies
Dhanya Vijayakumar, Joseph Jankovic
CNS Drugs.2022; 36(4): 327. CrossRef - Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases
Urmi Sengupta, Rakez Kayed
Progress in Neurobiology.2022; 214: 102270. CrossRef - Modeling the dynamics of innate immune response to Parkinson disease with therapeutic approach
Asma Badrah, Salma Al-Tuwairqi
Physical Biology.2022; 19(5): 056004. CrossRef - Potential of food-derived bioactive peptides in alleviation and prevention of Alzheimer's disease
Le Zhao, Dan Li, Xiaofen Qi, Kaifang Guan, Haoran Chen, Rongchun Wang, Ying Ma
Food & Function.2022; 13(21): 10851. CrossRef - Harnessing the immune system for the treatment of Parkinson’s disease
Vinata Vedam-Mai
Brain Research.2021; 1758: 147308. CrossRef - The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease
Katja Badanjak, Sonja Fixemer, Semra Smajić, Alexander Skupin, Anne Grünewald
International Journal of Molecular Sciences.2021; 22(9): 4676. CrossRef - Viral alpha-synuclein knockdown prevents spreading synucleinopathy
Sindhu Menon, Rikke H Kofoed, Fadl Nabbouh, Kristiana Xhima, Yasmeen Al-Fahoum, Tammy Langman, Howard T J Mount, Lamya S Shihabuddin, S Pablo Sardi, Paul E Fraser, Joel C Watts, Isabelle Aubert, Anurag Tandon
Brain Communications.2021;[Epub] CrossRef - Immunotherapies for Aging-Related Neurodegenerative Diseases—Emerging Perspectives and New Targets
Somin Kwon, Michiyo Iba, Changyoun Kim, Eliezer Masliah
Neurotherapeutics.2020; 17(3): 935. CrossRef - The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease
Kyu-Young Sim, Kyeong Chan Im, Sung-Gyoo Park
International Journal of Molecular Sciences.2020; 21(15): 5295. CrossRef - Novel antibodies detect additional α-synuclein pathology in synucleinopathies: potential development for immunotherapy
Jacqui T. Nimmo, Ajay Verma, Jean-Cosme Dodart, Chang Yi Wang, Jimmy Savistchenko, Ronald Melki, Roxana O. Carare, James A. R. Nicoll
Alzheimer's Research & Therapy.2020;[Epub] CrossRef - New Insights Into Drug Discovery Targeting Tau Protein
Yoshiyuki Soeda, Akihiko Takashima
Frontiers in Molecular Neuroscience.2020;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite