E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 12(3); 2019 > Article
-
Viewpoint
Recent Advances in the Development of Experimental Therapeutics for Levodopa-Induced Dyskinesia -
Michael L. Martini1,2
 , Sean N. Neifert1, J Mocco1, Fedor Panov1, Winona Tse3, Ruth H. Walker3,4, Jian Jin2, Fiona Gupta3
, Sean N. Neifert1, J Mocco1, Fedor Panov1, Winona Tse3, Ruth H. Walker3,4, Jian Jin2, Fiona Gupta3 -
Journal of Movement Disorders 2019;12(3):161-165.
DOI: https://doi.org/10.14802/jmd.19029
Published online: September 30, 2019
1Department of Neurosurgery, Icahn School of Medicine at Mount Sinai, New York, NY, USA
2Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
3Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
4James J. Peters VA Medical Center, Bronx, NY, USA
- Corresponding author: Michael L. Martini, BA Department of Neurosurgery, Icahn School of Medicine at Mount Sinai, 1 Gustave Levy Place, New York, NY 10029, USA / Tel: +1-212-659-8629 / E-mail: michael.martini@icahn.mssm.edu
Copyright © 2019 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide, with an estimated prevalence of approximately 1% in people over 60, and it represents an increasingly important medical problem in our aging population [1]. For decades, the standard of care for PD has involved treatment with levodopa (L-DOPA), which elevates dopamine levels in the nigrostriatal pathway, enhancing movement and coordinated motor functions. Chronic L-DOPA results in motor complications, including levodopa-induced dyskinesia (LID), which occur in at least 50% of patients after 5 to 10 years of treatment. LID is a significant limitation to the viability of long-term L-DOPA use because patient function and quality of life are compromised and individual and societal costs are increased [2].
- Although treating LID in PD patients can be challenging, significant progress has been made in the past few years. Experimental treatments are making their way through preclinical and clinical stages. This review will focus on data that demonstrate the rationale, safety, efficacy, and long-term benefits of these medical treatments in both animal models of PD and PD patients suffering from LID.
INTRODUCTION
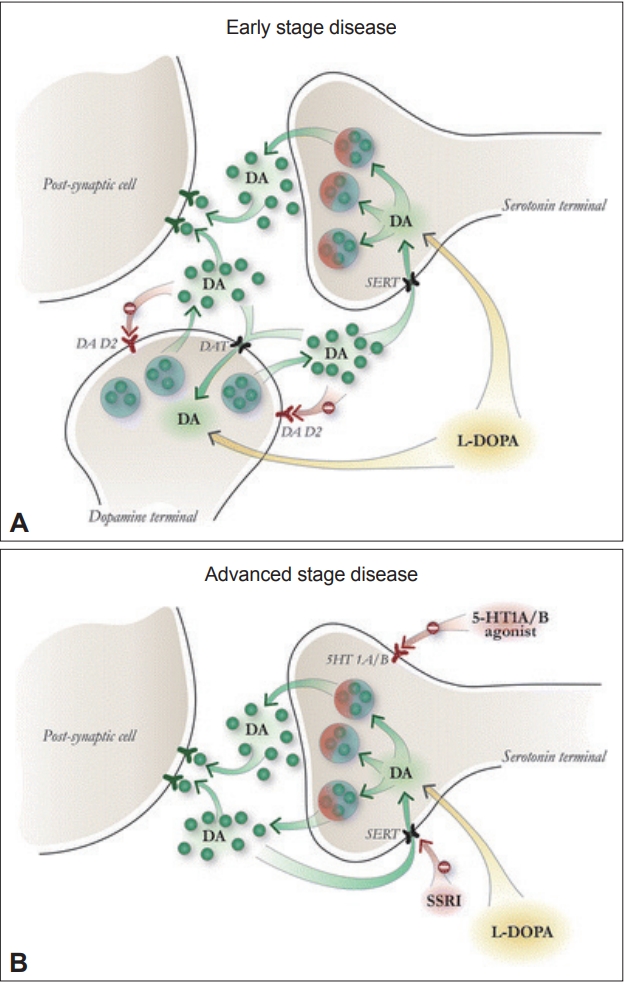
- Serotonergic neurons are a significant source of dopamine release in striatal synapses and possess the enzymes necessary to convert L-DOPA into dopamine [3]. When L-DOPA is administered to PD patients, synaptic dopamine levels oscillate as serotonergic neurons metabolize L-DOPA to dopamine but fail to autoregulate in response to elevated dopamine levels (Figure 1) [4]. PET studies have confirmed that L-DOPA produces higher synaptic dopamine levels in dyskinetic PD patients than in non-dyskinetic patients [5]. This loss of the ability to buffer excess synaptic dopamine is a proposed mechanism for LID development and may explain why LID is more prevalent when L-DOPA is administered orally than with continuous duodenal infusion.
- Pharmacological validation of this framework has been provided by several preclinical experiments. The administration of a selective 5-HT1 agonist to dampen serotonin neuron-derived dopamine release by autoreceptor stimulation significantly reduced LID in animals [6]. A synergistic effect on LID reduction was observed when 5-HT1A and 5-HT1B agonists were coadministered in rodent and primate [6] PD models. Selective serotonin reuptake inhibitors (SSRIs), including fluoxetine and citalopram, completely attenuated LID when they were administered to rodent models of PD, and this effect was blocked by the coadministration of a 5-HT1 antagonist [7]. Unlike 5-HT1 agonists, SSRIs do not reduce L-DOPA efficacy, suggesting that the temporarily reduced serotonin synaptic output from agonist-stimulated 5-HT1 autoreceptors may be responsible. This finding has additional clinical relevance when one considers that PD patients often experience depressive symptoms alongside motor fluctuations [8]. SSRIs may therefore be especially useful in these situations.
- Until recently, there was a dearth of clinical evidence supporting the idea that serotonergic neurons represent a viable therapeutic target for LID. Previously, a double-blinded, multicenter, open-label study found no improvement in LID in PD patients treated with the partial 5-HT1A agonist sarizotan [9,10]. This lack of efficacy was speculated to be due to the fact that sarizotan only selectively agonizes the 5-HT1A receptor and that the synergistic effect of 5-HT1B on LID reduction was not utilized. Promising results have emerged from a recent phase I/IIa randomized controlled trial for the mixed 5-HT1A/B agonist eltoprazine. Eltoprazine (5 and 7.5 mg) significantly reduced dyskinetic symptoms, as measured by both the Clinical Dyskinesia Rating Scale and the Rush Dyskinesia Rating Scale [11]. Importantly, Unified Parkinson’s Disease Rating Scale III scores did not differ significantly between the treatment and control groups, and patients treated with eltoprazine did not experience any decline in the therapeutic efficacy of subsequently administered L-DOPA. While the effects of long-term treatment with eltoprazine have not yet been established, early studies have shown very promising results, and further studies are underway to establish optimal dosing regimens in which the drug is delivered over extended periods of time.
- Together, preclinical and clinical data have demonstrated exciting advancements in understanding how perturbations in neurotransmitter systems can manifest as LID. These findings justify further studies with additional pharmacological agents that modulate the serotonergic system for the purpose of reducing LID.
SEROTONIN RECEPTORS
- In 2017, AT-390 and AT-403, two chemically distinct agonists of the nociceptin/orphanin FQ opioid peptide (NOP) receptor, were discovered [12]. These compounds were examined based on previous findings that NOP receptor agonists have the potential to decrease LID symptoms in animal models of PD [13]. Both compounds improved akinetic and dyskinetic symptoms in rodents, likely through an extracellular signal-regulated kinase (ERK)-related signaling mechanism and have the potential to be useful tools for further probing the role of NOP receptors in LID pathophysiology.
- Another recent pharmacological approach for attenuating LID involves targeting delta and mu opioid receptors. The stimulation of the delta opioid receptor improves PD symptoms in animals [14], while antagonizing mu opioid receptors diminishes LID symptoms [15]. A novel compound, termed DPI-289, was designed as a combined delta opioid receptor agonist/mu opioid receptor antagonist [16] and possesses antiparkinsonian and antidyskinetic properties in rodent and primate PD models. As a monotherapy in PD animal models, this compound exhibited significant, though limited, antiparkinsonian effects without any associated dyskinesia. In contrast, when DPI-289 was coadministered with L-DOPA, which is known to produce LID in these animals, a significant increase in anti-parkinsonian effects was observed, but it still did not produce any significant dyskinetic symptoms. These preclinical findings suggest that a drug with this polypharmacological profile may serve as a useful adjunctive therapy to traditional dopaminergic therapy for PD to alleviate motor deficits without eliciting associated LID symptoms.
OPIOID RECEPTOR MODULATORS
- Previous studies have shown that there is enhanced oxidative stress in the brain tissue of patients treated with L-DOPA and that this may be the result of decreased levels of antioxidants, the excessive oxidation of dopamine, and disruptions in the mitochondrial transport chain [17]. Additional studies have found that PD patients treated with L-DOPA have increased plasma levels of neuroinflammation markers, including oxidized-low density lipoproteins and soluble intracellular adhesion molecule [18]. Collectively, the literature suggests that monitoring oxidative stress markers and inflammatory factors may be useful for PD patients undergoing treatment with L-DOPA [19].
- Previous work has indicated that α-lipoic acid (ALA) can scavenge reactive oxygen species (ROS) and inhibit free radical generation via metal chelation [20]. These antioxidative processes suggest that ALA treatment may be useful for various disorders in which oxidative stress is proposed to play a role. Furthermore, ALA may also possess anti-inflammatory properties and may promote increases in intracellular glutathione formation, which has been postulated to be beneficial in neurodegenerative diseases [21]. In fact, previous studies have shown that ALA treatment attenuates ROS formation, resulting in neuroprotective effects on dopaminergic neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced apoptosis [22].
- Based on these purported pathophysiological processes and the proposed mechanism of action of ALA, a recent study supported the hypothesis that ALA may serve as a useful diseasemodifying therapy and attenuate LID when coadministered with L-DOPA in an animal model of PD [19]. This study by Zhang et al. [19] found that cotreatment had dose-dependent antidyskinetic effects. Molecular biomarkers and metabolites have been profiled in the substantia nigra, revealing that the antidegenerative effects of ALA likely stem from its ability to reduce oxidative stress and apoptosis in this neuronal cell population [19]. ALA may be a promising disease-modifying therapy that slows the loss of dopaminergic neurons in patients with early-stage PD and could allow for earlier incorporation of L-DOPA into medical regimens to significantly slow symptomatic disease progression while also delaying LID onset.
LIPOIC ACID
- It is hypothesized that the inhibition of glutamatergic N-methyl-D-aspartate (NMDA) receptors may be beneficial in PD since central dopamine depletion in the nigrostriatal pathway results in excessive glutamatergic activity in the basal ganglia [23]. Animal studies have indicated that selectively inhibiting a specific subset of NMDA receptors containing the NMDA Receptor 2B subunit (NR2B) subunit elicits a therapeutic effect and may potentiate the positive effects observed with dopaminergic therapy [24].
- Promising results have been observed in preclinical studies, but human studies have shown mixed results. A recent phase Ib randomized, double-blind, controlled crossover study was conducted to investigate the efficacy of a single dose of MK-0657, an NMDA receptor antagonist, against the motor symptoms of LID and PD [25]. The study found that MK-0657 did not significantly improve dyskinesia or motor function over a 5-hour period. Earlier studies conducted to assess the efficacy of CP-101,606, another NR2B-selective NMDA antagonist, however, did find significant beneficial effects on the motor symptoms of both LID [26] and PD [24,26].
- There may be a number of pharmacodynamic reasons for the inconsistencies among clinical trial results regarding different NR2B-selective NMDA antagonists. The research community still considers the NR2B subunit and other glutamatergic receptors to be valuable targets for LID drug discovery, given the robust findings implicating their pathological functions in key motor areas in PD patients. Several preclinical studies other modulators of metabotropic glutamate receptors for LID and PD treatment are still underway [27,28].
GLUTAMATERGIC RECEPTOR MODULATORS
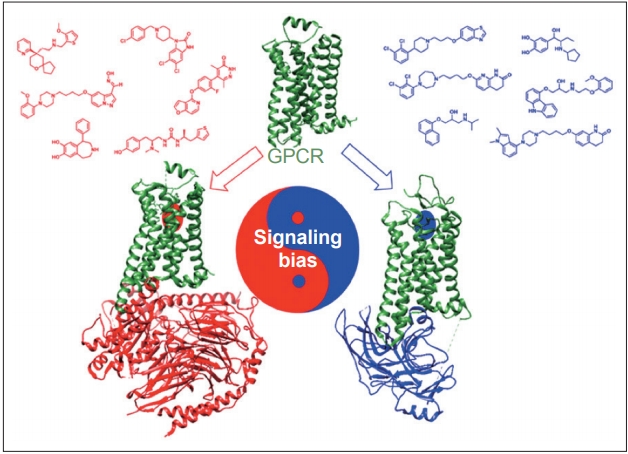
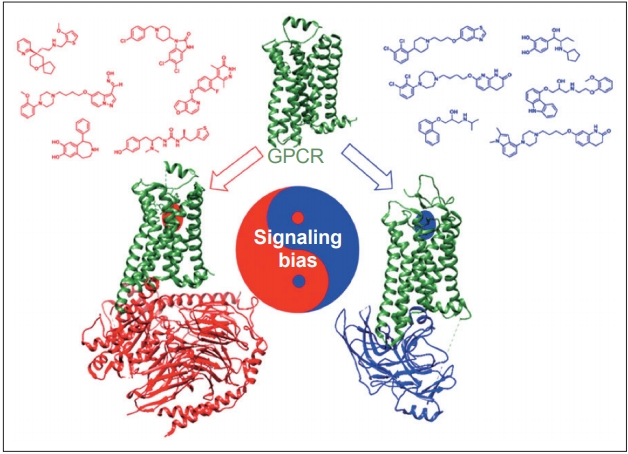
- Dopamine receptors are classified as G protein-coupled receptors (GPCRs), a superfamily of proteins that are targeted by approximately 25% of currently approved drugs [29]. A recent breakthrough in the GPCR pharmacology field is the ability to design drugs with functional selectivity, or signaling bias, whereby the GPCR ligand can selectively activate either G protein- or β-arrestin-mediated downstream signaling pathways (Figure 2) [30]. Functional selectivity offers the possibility to create drugs that can maintain the therapeutic benefits of their targets while dramatically decreasing the adverse side effect profiles that often come with targeting certain GPCRs [31]. One particularly promising application of this principle involves the possibility of treating the motor symptoms of PD without inducing LID.
- One of the prevailing hypotheses of LID involves GPCRs on dopaminergic neurons entering a supersensitive state after being deprived of postsynaptic dopamine due to PD. This supersensitive state results in enhanced G protein-mediated signaling [32], which is believed to contribute to uncontrolled downstream signaling and neuronal hyperactivity [33]. Several attempts, including attempts to decrease dopamine receptor surface expression on neurons [34], reduce abnormal signaling [35], and inhibit A2A [36], mGluR5 [27], or NMDA receptors [37], have been made to limit either uncontrolled signaling or neuronal hyperactivity. While none of these have proven successful, one promising strategy to address this problem involves the selective activation of β-arrestins, which act as signal transducers downstream of GPCRs [38] in motor-related signaling pathways. It has been hypothesized that selectively activating β-arrestin2 on dopamine receptors reduces LID by desensitizing supersensitive GPCRs while still maintaining the intracellular signaling that allows for improved motor function [33].
- Animal studies have supported the notion that LID is associated with increased G protein signaling and have also validated β-arrestins as novel targets for treating PD without causing LID. For example, knocking out β-arrestin2 in nonhuman primate models of PD resulted in worsened LID following L-DOPA administration, while β-arrestin2 overexpression reduced LID and increased locomotion via β-arrestin-mediated signaling [33].
- Although further preclinical and clinical testing must be done, these results have generated significant interest in this area. Recently, Gray et al. [39] reported a novel noncatechol-containing agonist scaffold with excellent blood-brain barrier permeability, potency, and selectivity for the D1 dopamine receptor (D1R). Although the reported scaffold is highly biased for the stimulatory G protein (GS) pathway, displaying almost no activation of β-arrestin2 recruitment to D1R, the scaffold has been shown to have a sustained effect during a 3-day period on eye-blink rate in a nonhuman primate model of PD, which is considered a functional marker of central dopaminergic activity mediated through D1R [39]. The sustained response observed in this short-term study was attributed to reduced desensitization and tachyphylaxis associated with diminished β-arrestin2 signaling. Although a fully β-arrestin2-biased D1R ligand has not yet been reported, recent studies have reported promising findings regarding adapting the novel noncatechol D1R scaffold to increase its β-arrestin2 bias [40]. Advancements in GPCR drug discovery and biological validation have ensured that progress towards developing a functionally selective clinical drug candidate for the treatment of PD is underway.
β-ARRESTINS
- L-DOPA is a staple of therapeutic regimens for PD patients because it effectively improves motor function and quality of life. LID emerges after chronic L-DOPA use and often complicates therapeutic management. Recent innovations have spawned new pharmacological strategies for the treatment of LID, and these innovations have progressed significantly in recent years and have shown great promise in preclinical and clinical studies. It is possible that some of these strategies may eventually be a viable option for patients who are ineligible for neurosurgical procedures or unable to tolerate the adverse side effects of other pharmacological interventions.
CONCLUSION
- This work was supported in part by the F30AG062054 from the U.S. National Institutes of Health, awarded to M.L.M.
Acknowledgments

- 1. de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol 2006;5:525–535.ArticlePubMed
- 2. Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson’s disease (2001): treatment guidelines. Neurology 2001;56(11 Suppl 5):S1–S88.Article
- 3. Nevalainen N, Af Bjerkén S, Gerhardt GA, Strömberg I. Serotonergic nerve fibers in L-DOPA-derived dopamine release and dyskinesia. Neuroscience 2014;260:73–86.ArticlePubMed
- 4. Carta M, Björklund A. The serotonergic system in L-DOPA-induced dyskinesia: pre-clinical evidence and clinical perspective. J Neural Transm (Vienna) 2018;125:1195–1202.ArticlePubMedPDF
- 5. Pagano G, Yousaf T, Politis M. PET molecular imaging research of levodopa-induced dyskinesias in Parkinson’s disease. Curr Neurol Neurosci Rep 2017;17:90.ArticlePubMedPMCPDF
- 6. Muñoz A, Li Q, Gardoni F, Marcello E, Qin C, Carlsson T, et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 2008;131(Pt 12):3380–3394.ArticlePubMedPDF
- 7. Conti MM, Ostock CY, Lindenbach D, Goldenberg AA, Kampton E, Dell’isola R, et al. Effects of prolonged selective serotonin reuptake inhibition on the development and expression of L-DOPA-induced dyskinesia in hemi-parkinsonian rats. Neuropharmacology 2014;77:1–8.ArticlePubMed
- 8. Marsh L. Depression and Parkinson’s disease: current knowledge. Curr Neurol Neurosci Rep 2013;13:409.ArticlePubMedPMCPDF
- 9. Goetz CG, Damier P, Hicking C, Laska E, Müller T, Olanow CW, et al. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov Disord 2007;22:179–186.ArticlePubMed
- 10. Olanow CW, Damier P, Goetz CG, Mueller T, Nutt J, Rascol O, et al. Multicenter, open-label, trial of sarizotan in Parkinson disease patients with levodopa-induced dyskinesias (the SPLENDID Study). Clin Neuropharmacol 2004;27:58–62.ArticlePubMed
- 11. Svenningsson P, Rosenblad C, Af Edholm Arvidsson K, Wictorin K, Keywood C, Shankar B, et al. Eltoprazine counteracts l-DOPA-induced dyskinesias in Parkinson’s disease: a dose-finding study. Brain 2015;138(Pt 4):963–973.ArticlePubMedPMCPDF
- 12. Arcuri L, Novello S, Frassineti M, Mercatelli D, Pisanò CA, Morella I, et al. Anti-Parkinsonian and anti-dyskinetic profiles of two novel potent and selective nociceptin/orphanin FQ receptor agonists. Br J Pharmacol 2018;175:782–796.ArticlePubMedPMC
- 13. Marti M, Rodi D, Li Q, Guerrini R, Fasano S, Morella I, et al. Nociceptin/orphanin FQ receptor agonists attenuate L-DOPA-induced dyskinesias. J Neurosci 2012;32:16106–16119.ArticlePubMedPMC
- 14. Hille CJ, Fox SH, Maneuf YP, Crossman AR, Brotchie JM. Antiparkinsonian action of a delta opioid agonist in rodent and primate models of Parkinson’s disease. Exp Neurol 2001;172:189–198.ArticlePubMed
- 15. Koprich JB, Fox SH, Johnston TH, Goodman A, Le Bourdonnec B, Dolle RE, et al. The selective mu-opioid receptor antagonist ADL5510 reduces levodopa-induced dyskinesia without affecting antiparkinsonian action in MPTP-lesioned macaque model of Parkinson’s disease. Mov Disord 2011;26:1225–1233.ArticlePubMed
- 16. Johnston TH, Versi E, Howson PA, Ravenscroft P, Fox SH, Hill MP, et al. DPI-289, a novel mixed delta opioid agonist / mu opioid antagonist (DAMA), has L-DOPA-sparing potential in Parkinson’s disease. Neuropharmacology 2018;131:116–127.ArticlePubMed
- 17. Spencer JP, Jenner P, Halliwell B. Superoxide-dependent depletion of reduced glutathione by L-DOPA and dopamine. Relevance to Parkinson’s disease. Neuroreport 1995;6:1480–1484.ArticlePubMed
- 18. Andican G, Konukoglu D, Bozluolcay M, Bayülkem K, Firtiına S, Burcak G. Plasma oxidative and inflammatory markers in patients with idiopathic Parkinson’s disease. Acta Neurol Belg 2012;112:155–159.ArticlePubMedPDF
- 19. Zhang SF, Xie CL, Lin JY, Wang MH, Wang XJ, Liu ZG. Lipoic acid alleviates L-DOPA-induced dyskinesia in 6-OHDA parkinsonian rats via anti-oxidative stress. Mol Med Rep 2018;17:1118–1124.ArticlePubMed
- 20. Smith AR, Shenvi SV, Widlansky M, Suh JH, Hagen TM. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem 2004;11:1135–1146.ArticlePubMed
- 21. De Araújo DP, Lobato Rde F, Cavalcanti JR, Sampaio LR, Araújo PV, Silva MC, et al. The contributions of antioxidant activity of lipoic acid in reducing neurogenerative progression of Parkinson’s disease: a review. Int J Neurosci 2011;121:51–57.ArticlePubMed
- 22. Li DW, Li GR, Lu Y, Liu ZQ, Chang M, Yao M, et al. α-lipoic acid protects dopaminergic neurons against MPP+-induced apoptosis by attenuating reactive oxygen species formation. Int J Mol Med 2013;32:108–114.ArticlePubMed
- 23. Chase TN, Oh JD, Konitsiotis S. Antiparkinsonian and antidyskinetic activity of drugs targeting central glutamatergic mechanisms. J Neurol 2000;247 Suppl 2:II36–II42.ArticlePubMedPDF
- 24. Steece-Collier K, Chambers LK, Jaw-Tsai SS, Menniti FS, Greenamyre JT. Antiparkinsonian actions of CP-101,606, an antagonist of NR2B subunit-containing N-methyl-d-aspartate receptors. Exp Neurol 2000;163:239–243.ArticlePubMed
- 25. Herring WJ, Assaid C, Budd K, Vargo R, Mazenko RS, Lines C, et al. A phase Ib randomized controlled study to evaluate the effectiveness of a single-dose of the NR2B selective N-methyl-D-aspartate antagonist MK-0657 on levodopa-induced dyskinesias and motor symptoms in patients with Parkinson disease. Clin Neuropharmacol 2017;40:255–260.ArticlePubMed
- 26. Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M, et al. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord 2008;23:1860–1866.ArticlePubMedPMC
- 27. Rascol O, Fox S, Gasparini F, Kenney C, Di Paolo T, Gomez-Mancilla B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat Disord 2014;20:947–956.ArticlePubMed
- 28. Litim N, Morissette M, Di Paolo T. Metabotropic glutamate receptors as therapeutic targets in Parkinson’s disease: an update from the last 5 years of research. Neuropharmacology 2017;115:166–179.ArticlePubMed
- 29. Wacker D, Stevens RC, Roth BL. How ligands illuminate GPCR molecular pharmacology. Cell 2017;170:414–427.ArticlePubMedPMC
- 30. Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 2014;35:308–316.ArticlePubMed
- 31. Tan L, Yan W, McCorvy JD, Cheng J. Biased ligands of G protein-coupled receptors (GPCRs): structure-functional selectivity relationships (SFSRs) and therapeutic potential. J Med Chem 2018;61:9841–9878.ArticlePubMed
- 32. Fiorentini C, Savoia P, Savoldi D, Barbon A, Missale C. Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Neurobiol Dis 2013;54:339–348.ArticlePubMed
- 33. Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, et al. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc Natl Acad Sci U S A 2015;112:E2517–E2526.ArticlePubMedPMC
- 34. Porras G, Berthet A, Dehay B, Li Q, Ladepeche L, Normand E, et al. PSD-95 expression controls L-DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest 2012;122:3977–3989.ArticlePubMedPMC
- 35. Feyder M, Södersten E, Santini E, Vialou V, LaPlant Q, Watts EL, et al. A role for mitogen- and stress-activated kinase 1 in L-DOPA-induced dyskinesia and ΔFosB expression. Biol Psychiatry 2016;79:362–371.ArticlePubMed
- 36. Mizuno Y, Hasegawa K, Kondo T, Kuno S, Yamamoto M; Japanese Istradefylline Study Group. Clinical efficacy of istradefylline (KW-6002) in Parkinson’s disease: a randomized, controlled study. Mov Disord 2010;25:1437–1443.ArticlePubMed
- 37. Ory-Magne F, Corvol JC, Azulay JP, Bonnet AM, Brefel-Courbon C, Damier P, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology 2014;82:300–307.ArticlePubMed
- 38. Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A 2001;98:2449–2454.ArticlePubMedPMC
- 39. Gray DL, Allen JA, Mente S, O’Connor RE, DeMarco GJ, Efremov I, et al. Impaired β-arrestin recruitment and reduced desensitization by noncatechol agonists of the D1 dopamine receptor. Nat Commun 2018;9:674.ArticlePubMedPMCPDF
- 40. Martini ML, Liu J, Ray C, Yu X, Huang XP, Urs A, et al. Defining structure-functional selectivity relationships (SFSR) for a class of non-catechol dopamine D1 receptor agonists. J Med Chem 2019;62:3753–3772.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Antioxidant Effect of Alpha-Lipoic Acid in 6-Hydroxydopamine Unilateral Intrastriatal Injected Rats
Pavlina Andreeva-Gateva, Lubomir Traikov, Zafer Sabit, Dimitar Bakalov, Radka Tafradjiiska-Hadjiolova
Antioxidants.2020; 9(2): 122. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite