E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 12(3); 2019 > Article
-
Original Article
Clinical Milestones Preceding the Diagnosis of Multiple System Atrophy and Progressive Supranuclear Palsy: A Retrospective Cohort Study -
Louise Wiblin1
 , Rory Durcan2, Brook Galna2, Mark Lee3, David Burn2
, Rory Durcan2, Brook Galna2, Mark Lee3, David Burn2 -
Journal of Movement Disorders 2019;12(3):177-183.
DOI: https://doi.org/10.14802/jmd.19015
Published online: August 9, 2019
1Department of Neurosciences, Royal Victoria Infirmary, Newcastle-upon-Tyne, UK
2Institute of Neuroscience, Newcastle University, Newcastle-upon-Tyne, UK
3St. Benedict’s Hospice, Sunderland, UK
- Corresponding author: Louise Wiblin, MRCP, MD Department of Neurosciences, Royal Victoria Infirmary, Queen Victoria Rd, Newcastle-upon-Tyne NE1 4LP, UK / Tel: +44-0191-233-6161 / E-mail: l.wiblin@nhs.net
Copyright © 2019 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Objective
- Multiple System Atrophy (MSA) and progressive supranuclear palsy (PSP) are rapidly progressive forms of degenerative Parkinsonism. The difficulties of diagnosing MSA and PSP in their early stages may lead to delayed referral to appropriate specialists and distress to patients, as well as delaying symptomatic treatment and participation in clinical trials. This work aimed to describe the symptoms that patients with MSA and PSP developed and plot their emergence relative to final diagnosis using a median onset in months.
-
Methods
- Forty-seven patients from the United Kingdom with MSA or PSP diagnosed by a movement disorder specialist were interviewed with carers or relatives to establish milestone onset. This was corroborated using clinical notes and letters.
-
Results
- In the MSA cohort (n = 23), autonomic symptoms (median 5.5 months before diagnosis) and falls (median 1 month before diagnosis) were the two clinical milestones which occurred before diagnosis. In the PSP cohort (n = 24), falling was the only milestone which occurred before diagnosis (median of 18.5 months).
-
Conclusion
- This Study Shows That Psp Patients Experience Falling More Than A Year And A Half An Average Before Receiving A Diagnosis And Although Msa Patients Also Tended To Fall, This Was Much Closer To The Time Of Diagnosis. Further Work With Larger Cohorts May Illustrate Whether These Preliminary Findings Can Be Generalised To Guide Diagnosis And Management.
- Patient recruitment and inclusion
- Forty-seven patients were recruited from the north-east of England, 23 with a diagnosis of MSA and 24 with PSP. All patients were diagnosed by a neurologist specializing in movement disorders and fulfilled the Second Consensus Criteria for the diagnosis of MSA or the NINDS-SPSP criteria for PSP where appropriate [9,10]. The Höglinger Criteria for PSP was released after the close of the study but was imposed afterwards for consistency to check the accuracy of diagnosis, as the Höglinger Criteria are more sensitive and specific for progressive supranuclear palsy-Parkinsonism (PSP-P) and other forms such as pure akinesia with gait freezing (PAGF) (Table 1 and 2) [5]. The study aimed to recruit a minimum of 20 patients with MSA and 20 with PSP as part of a wider study looking at the trajectory of diagnosis, symptom onset and a number of other variables such as quality of life. There was no upper limit for recruitment. Sample size was determined on the basis of obtaining meaningful pilot data and the population of patients within the geographical region [11]. Patients were approached by their neurologist within the movement disorder clinic in Newcastle, Sunderland or Middlesbrough in the UK, which were participant recruitment centers. It was made explicit to all patients that electing not to take part would have no impact upon their clinical care.
- Data collection
- Ten key clinical milestones were selected after a review of the literature (Table 3 and 4) [1]. These were the onset of falls, autonomic dysfunction (here defined as orthostatic hypotension and urinary dysfunction), the need for a wheelchair, speech problems, swallowing difficulty, acute admission as a consequence of atypical Parkinsonism, catheterization, PEG insertion, tracheostomy and admission to a care facility. These milestones were selected on the basis of clinical utility. They reflect important markers of disease progression, tend to be noticeable when they occur, are readily recalled by the patient or carer, and thus are more likely to be documented in medical notes or clinical letters. These milestones were defined using the same wording and definitions for consistency between participants (e.g., definition of dysarthria was ‘the point at which it was felt that you were difficult to understand by others’). The median time to onset for each milestone within the MSA and PSP groups was calculated relative to the time of diagnosis.
- If a participant reached a milestone that was attributable to another established condition or cause, they were excluded from the analysis of that clinical milestone only. This was the case for one participant with MSA who received a catheter following surgery for prostate cancer 26 years prior to the interview and a participant with PSP who used a wheelchair due to a spinal injury incurred 11 years before the diagnosis of PSP.
- Analysis
- Milestone-onset was corroborated by the carer/relative, medical notes and clinical letters. All participants’ interviews were cross-checked with their notes and clinical letters, and all but one patient in the PSP group had a carer to corroborate patient accounts (this carer planned to attend the research facility but failed to do so). The onset of each milestone was recorded in months and compared with the date of diagnosis with MSA or PSP (for consistency, the time of diagnosis was defined as the revised diagnosis of MSA or PSP rather than an earlier misdiagnosis of PD, for example).
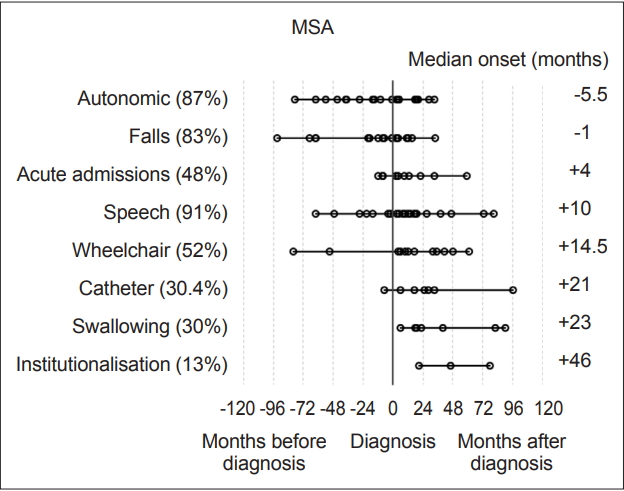
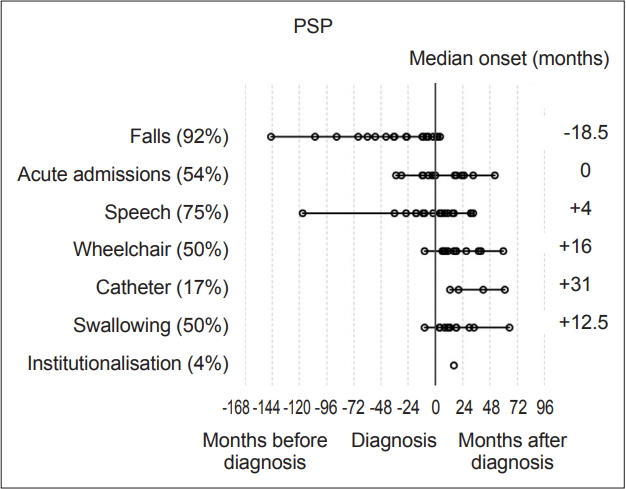
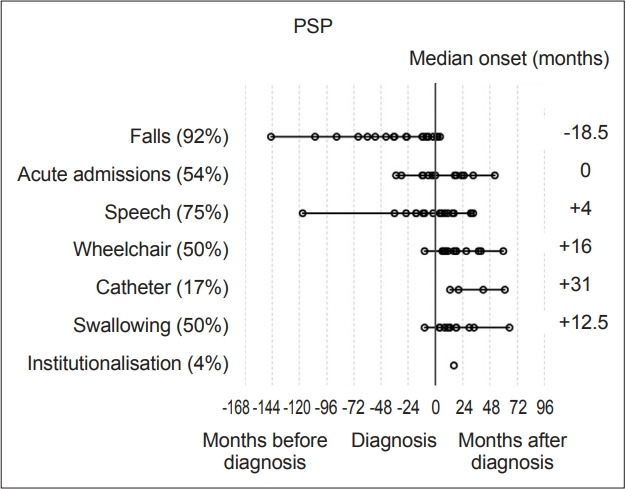
- Statistical analysis was carried out with IBM SPSS version 23 (IBM Corp., Armonk, NY, USA). Milestones, such as speech problems, were reviewed and plotted on graphs to visually represent their onset relative to diagnosis (‘-’ numbers signifying before diagnosis and ‘+’ after) Each milestone was then analyzed within disease groups for its median-onset relative to the point of diagnosis. These values were then plotted to determine the temporal sequence of onset compared to other milestones and the time of diagnosis (Figure 1 and 2). Median scores were used to describe these onset times as their distributions were not normal on visual inspection of histograms and on the basis of the results of Shapiro-Wilk tests.
- Ethical statement
- All procedures performed in studies involving human participants were conducted in accordance with the ethical standards of Newcastle University and the Humber-Leeds/Bradford branch of the UK Ethics Committee. These assess the compliance of studies with the 1975 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all the patients included in the study. Permissions were given by Yorkshire and the Humber-Leeds/Bradford Regional Ethics Committee (REC), study number 15/YH/0459.
MATERIALS & METHODS
- Of the 47 patients, 23 had received a prior diagnosis before it was revised to MSA or PSP. In the MSA group, 11 out of 23 had a prior diagnosis (10 MSA-P, 1 MSA-C), all of which were PD. The median time to the revision of the diagnosis was 41 months. In the PSP group, 12 out of 24 had a prior diagnosis. Nine patients were originally diagnosed with PD, 7 of which were later revised to PSP-P. The remaining 2 were rediagnosed as having progressive supranuclear palsy-Richardson’s syndrome (PSPRS). Three patients received prior diagnoses other than PD: frontotemporal dementia, ‘mixed dementia’ and ‘stroke disease’ These latter three patients were all rediagnosed as having PSPRS; however, it should be noted that in those cases, the original diagnoses were not made by a neurologist or movement disorder specialist. The median time to rediagnosis in the PSP group was also a median of 41 months.
- Of the 23 MSA patients, 11 were male and 12 were female. The mean age for patients with MSA was 64.6 years (SD 10.1), and the median disease duration was 33 months (IQR: 29 months, range 5–105 months). Sixteen patients had MSA-P, and 7 had MSA-C. Using the Second Consensus Criteria, 17 patients had probable MSA, and 6 had possible MSA [9].
- Of the 24 patients with PSP, 10 were female and 14 were male. The mean age was 71.6 years (SD 6.8), and the median disease duration was 25.5 months (IQR 30.8, range 9–215 months). Of the 24 patients with PSP, 13 had PSP-RS, 10 had PSP-P and one had PSP-PAGF. Using the NINDS-SPSP Criteria in use at the time of the study, 16 patients had probable PSP, and 7 had possible PSP [the remaining patient had progressive supranuclear palsy-progressive akinesia with gait freezing (PSP-PAGF), which is not well identified using the NINDS criteria]. After the release of the Höglinger Criteria, the examination findings were compared: 19 had probable PSP and 5 had possible PSP. The improvement is likely due to the better detection of subtypes such as PSP-P and PAGF using these new criteria.
- Overall, the most prevalent milestones in MSA were autonomic symptoms (20/23, 87%), speech disturbance (21/23, 91%) and falls (19/23, 83%). Three patients (13%) denied autonomic symptoms; however, one patient had well-documented erectile dysfunction and urinary incontinence, although during the research consultation, they declined to discuss or acknowledge this, likely due to embarrassment. The remaining two patients had objective abnormalities on autonomic function tests though they denied experiencing any subjective symptoms. Therefore, all MSA participants had autonomic dysfunction. Meanwhile, among the patients with PSP, the most prevalent milestones were falls (22/24, 92%), speech disturbance (18/24, 75%) and acute admissions (13/24, 54%).
- The most frequently occurring first milestones in patients with MSA were falls and autonomic dysfunction, with 10 patients reporting falls as their first milestone and 10 patients reporting autonomic symptoms as their first milestone (10/23, 43%); one patient had falls and autonomic symptoms with simultaneous onset as a joint first milestone. Speech disturbance was the first reported milestone in five patients in the MSA group (in one case reported at the same time as autonomic symptoms and in another at the same time as falls), while three PSP patients reported speech disturbance as their first (or joint first) milestone. Overall, the prevalence of speech disturbance was high among both the MSA (91.3%) and PSP groups (75%). The predominant first milestone in patients with PSP was falling: 19 of the 24 (79%) patients had falling as their first clinical milestone. Two patients reported speech disturbance as the first symptom noted, and 1 patient had speech disturbance and falls with onset at the same time (13%).
- The milestones with a median time of onset preceding a diagnosis of MSA were autonomic symptoms [median onset -5.5 months (range -79 to 33 months), IQR 55.5] and falls [-1 month (range -93 to 34 months), IQR 30.0].
- In patients with PSP, only falls had a median time of onset preceding a diagnosis, at -18.5 months [(range -145 to 4 months), IQR 49.8]. Need for a wheelchair occurred at a median of 12 months (range 2 to 113 months, IQR 50.3) after diagnosis in patients with MSA and 16 months (range 1 to 132 months, IQR 20.5) in patients with PSP (Figure 1 and 2).
- In terms of falling, when patients with MSA were split into subgroups, the MSA-P group fell a median of 3 months after diagnosis, whereas the MSA-C group fell a median of 19 months prior to diagnosis. There were no significant differences between the subtypes according to the Mann-Whitney U test (p = 0.097). The reason for this may be greater unsteadiness in the MSA-C group due to added ataxia combined with autonomic dysfunction. However, it is possible that the MSA-C group was diagnosed later after the onset of symptoms because the cerebellar form of MSA is less common in European populations, therefore experiencing a comparatively greater lag in reaching a movement disorder specialist [12].
- When the PSP group was divided into subtypes, the PSP-RS group fell a median of 36 months prior to diagnosis, whereas the PSP-P group fell a median of 7 months prior to diagnosis. There was no significant difference between the groups according to the Mann-Whitney U test (p = 0.071), which may be due to the small sample size. An earlier onset of falling in the RS subgroup of PSP would be expected from previous work suggesting a more rapid decline in the RS subtype [13].
- The time difference between the onset of the first milestone and diagnosis was compared using Mood’s Median test between the two disease groups, and there were no significant differences. Furthermore, there were no differences overall in the time difference from the onset of the first milestone to diagnosis among the three groups; with autonomic dysfunction as the first milestone, speech as the first milestone, and falls as the first milestone, according to Kruskal Wallis tests.
RESULTS
- This pilot study has some important implications, both for research purposes and in terms of clinical considerations. The ability to predict the disease type more accurately in the prediagnostic phase may have implications for earlier diagnosis, effective referral (which also helps patient uncertainty, a known burden), palliative care and ACP [14]. Identifying patient cohorts in the early stages of disease who are eligible for novel drug trials is also increasingly important. In this discussion, the findings of the study are considered in light of the possible implications and the work that needs to be done in the future.
- The first milestone perceived by MSA patients in this cohort before diagnosis was autonomic dysfunction, specifically orthostatic hypotension and urinary symptoms, with a median onset of 5.5 months prior to diagnosis. Falls were the only other milestone that had a median onset prior to diagnosis (1 month). Speech problems, autonomic symptoms and then falling were the most common milestones reached overall. In the PSP group, falls were the only milestone with a median onset prior to diagnosis at -18.5 months. In the PSP group, the three milestones with the highest overall incidence were falls, speech dysfunction and acute admissions, largely due to injuries sustained during falling.
- What are the ramifications of these findings and how might they relate to the issues surrounding MSA and PSP in practice? One of the current challenges posed by MSA and PSP is accurately diagnosis in the early stages; this encompasses not only the specialist differentiating between types of atypical parkinsonism but also general practitioners being able to identify patients needing a referral to a movement disorder specialist. It is these generalists who see the patients in the prediagnostic phase, and knowing which symptoms patients become aware of early on may allow us to guide generalists to refer these patients appropriately for a more rapid diagnosis.
- This pilot work also describes the onset of certain symptoms, such as dysphagia and speech difficulty, relative to the time of diagnosis. Having an idea of average disease trajectories is valuable to the clinician caring for these patients and their families. It can allow frank discussions and planning, incorporating ACP. Many patients wish to know how their disease will progress, and when they might expect particular issues to occur, such as loss of mobility. This can have implications for whether they can remain at home and which adaptations may be required to permit this.
- In addition to practical planning, a key part of caring for patients with neurodegenerative conditions such as MSA and PSP is discussing issues such as resuscitation and which treatments would be acceptable to patients while they are still able to communicate intelligibly. Having a guide as to when this may become problematic for the patient is helpful.
- Although this is a small sample population, and the clinical milestones were determined largely from retrospective history (albeit with corroboration from clinical notes and letters when possible), the onset of memorable symptoms and how they relate to the time of diagnosis is clinically useful. For example, if a patient with atypical parkinsonism (type not yet clear) has sustained falls for a year or more (not clearly related to hypotension), then according to the results of this study, it might be inferred that the patient will subsequently be diagnosed with PSP. The subjective perspective of patients and carers may also be helpful, as individuals often detect problems with normal functioning before they are ascribed to a unifying problem by clinicians.
- The limitations of this work are the small sample size, possibilities of recall bias on the part of participants, especially if reliable clinical documentation dating certain milestones was absent, and the cross-sectional nature of the study. Unfortunately, contemporaneous data collection of symptoms and signs prior to a rare diagnosis are unlikely, as population studies would not contain enough subjects likely to develop PSP or MSA. Large longitudinal studies are being conducted, but symptoms occurring before a diagnosis is made must be determined retrospectively. All participants were under the care of a specialist movement disorder neurologist, which adds bias; undiagnosed patients with MSA and PSP or patients who may be receiving care from nonspecialists and have not been referred to the movement disorder specialist services may well have a longer lag time to diagnosis, but these patients could not be easily identified within the limitations of the recruitment process. Patients were diagnosed according to the recommended criteria, but the gold standard of diagnosis in these conditions is an autopsy postmortem, which was beyond the scope of this study. Although the range of disease duration was large, the median disease duration was fairly short. The strengths of this study are its inclusiveness, the use of multiple sources to try and verify the timing of symptoms and the presentation of symptom trajectories relative to the point at which the patient was diagnosed; this study was a pragmatic attempt to delineate what patients experience before they achieve a diagnosis of atypical parkinsonism and how we may use this knowledge in the future to diagnose these patients earlier.
- There are large cohorts with MSA and PSP in observational studies, such as the PROgressive Supranuclear Palsy CorTico-Basal Syndrome Multiple System Atrophy Longitudinal Study UK (PROSPECT) study being conducted in the UK, which are exploring prediagnostic symptoms and will also follow patients over a number of years to produce a trajectory of disease progression [15]. Larger studies such as these may verify our findings of falls as a key prediagnostic symptom, with falls in PSP occurring more distantly from the time of diagnosis to MSA.
- This is a small pilot study that has shown that in a cohort of 47 patients with atypical parkinsonism, the MSA group of 23 patients had two key symptom milestones that arose before diagnosis: falls (1 month before) and autonomic symptoms (5.5 months before). In the PSP group (24 patients), only one milestone on average predated diagnosis, namely, falls, which occurred an average of 18.5 months before diagnosis.
- There may be a significant finding here that could be helpful in discriminating between MSA and PSP in the early phase of disease. This is important not only for addressing patient uncertainty and clinical planning for the future but also in selecting patients with early disease for trials and eventually, potentially disease-modifying treatments, as research continues to seek agents that are neuroprotective or interfere with disease progression.
DISCUSSION
-
Conflicts of Interest
None of the authors have any financial disclosures to disclose related to this work. Louise Wiblin, Rory Durcan and David Burn were involved in patient recruitment and data collection for the PROSPECT study cited in the references, although this study is not connected to that work. Otherwise, there are no declarations of conflicts of interest.
Notes

| Characteristics of the PSP group | |
|---|---|
| Number | 24 |
| Sex (female) | 14 |
| Age (years) | 71.6 (SD 6.8) |
| Subtype (n) | |
| PSP-RS | 13 |
| PSP-P | 10 |
| PSP-PAGF | 1 |
| NINDS-SPSP (1996) [10]/Höglinger (2017) [5] (n) | |
| Probable | 16/19 |
| Possible | 7/5 |
| Median duration of disease (months) | 25.5 (IQR 30.8) overall |
| PSP-RS | 21.0 (IQR 53.0) |
| PSP-P | 34.0 (IQR 59.0) |
| PSP-PAGF | 51.0 (IQR -) |
| Median duration of follow-up (months) | 51.0 (IQR 37.0) |
| PSP-RS | 49 (IQR 49.5) |
| PSP-P | 56.5 (IQR 48.8) |
| PSP-PAGF | - |
PSP: progressive supranuclear palsy, NINDS-SPSP: National Institute of Neurological Disorders and Stroke and the Society for PSP criteria, IQR: interquartile range, PSP-RS: progressive supranuclear palsy-Richardson’s syndrome, PSP-P: progressive supranuclear palsy-Parkinsonism, PSP-PAGF: progressive supranuclear palsy-progressive akinesia with gait freezing.
- 1. O’Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, et al. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain 2008;131(Pt 5):1362–1372.ArticlePubMedPDF
- 2. Jecmenica-Lukic M, Petrovic IN, Pekmezovic T, Kostic VS. Clinical outcomes of two main variants of progressive supranuclear palsy and multiple system atrophy: a prospective natural history study. J Neurol 2014;261:1575–1583.ArticlePubMedPDF
- 3. Coon EA, Sletten DM, Suarez MD, Mandrekar JN, Ahlskog JE, Bower JH, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain 2015;138(Pt 12):3623–3631.ArticlePubMedPMCPDF
- 4. Ling H. Clinical approach to progressive supranuclear palsy. J Mov Disord 2016;9:3–13.ArticlePubMedPMCPDF
- 5. Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 2017;32:853–864.ArticlePubMedPMC
- 6. dell’Aquila C, Zoccolella S, Cardinali V, de Mari M, Iliceto G, Tartaglione B, et al. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism Relat Disord 2013;19:980–985.ArticlePubMed
- 7. Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017;88:402–411.ArticlePubMed
- 8. Brooks DJ. Diagnosis and management of atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2002;72 Suppl 1:I10–I16.ArticlePubMed
- 9. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 10. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDSSPSP international workshop. Neurology 1996;47:1–9.ArticlePubMed
- 11. Hertzog MA. Considerations in determining sample size for pilot studies. Res Nurs Health 2008;31:180–191.ArticlePubMed
- 12. Ozawa T, Revesz T, Paviour D, Lees AJ, Quinn N, Tada M, et al. Difference in MSA phenotype distribution between populations: genetics or environment? J Parkinsons Dis 2012;2:7–18.ArticlePubMed
- 13. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSPparkinsonism. Brain 2005;128(Pt 6):1247–1258.ArticlePubMedPDF
- 14. Etkind SN, Koffman J. Approaches to managing uncertainty in people with life-limiting conditions: role of communication and palliative care. Postgrad Med J 2016;92:412–417.ArticlePubMed
- 15. Woodside J, Lamb R, Chelban V, Burn D, Church A, Gerhard A, et al. PROSPECT: a UK-based longitudinal observational study of PSP, CBD, MSA and atypical parkinsonism syndromes [abstract]. Mov Disord 2016;31(Suppl 2):A257.Article
REFERENCES
Figure & Data
References
Citations
- Clinical milestones as triggers for palliative care intervention in progressive Supranuclear palsy and multiple system atrophy
Robin Bessemer, Alla Iansavichene, Mary E. Jenkins, Elizabeth Finger, Teneille E. Gofton
Journal of the Neurological Sciences.2023; 448: 120614. CrossRef - Toward More Accessible Fully Automated 3D Volumetric MRI Decision Trees for the Differential Diagnosis of Multiple System Atrophy, Related Disorders, and Age-Matched Healthy Subjects
Jisoo Kim, Geoffrey S. Young, Andrew S. Willett, Ariana T. Pitaro, Grace F. Crotty, Merlyne Mesidor, Kristie A. Jones, Camden Bay, Min Zhang, Mel B. Feany, Xiaoyin Xu, Lei Qin, Vikram Khurana
The Cerebellum.2022; 22(6): 1098. CrossRef - Disease course and treatment patterns in progressive supranuclear palsy: A real-world study
John C. Morgan, Xiaolan Ye, Jennifer A. Mellor, Keisha J. Golden, Jorge Zamudio, Louis A. Chiodo, Yanjun Bao, Tao Xie
Journal of the Neurological Sciences.2021; 421: 117293. CrossRef - Patient and care partner views on exercise and structured physical activity for people with Progressive Supranuclear Palsy
Susan C. Slade, Christopher Bruce, Jennifer L. McGinley, Bastiaan R. Bloem, Meg E. Morris, John Duda
PLOS ONE.2020; 15(6): e0234265. CrossRef - Effect of cold oral stimulation on orthostatic hypotension in multiple system atrophy: a case study
Hironobu Uzawa, Shinta Takeuchi, Yusuke Nishida
Journal of Physical Therapy Science.2020; 32(7): 473. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite