Altered Gut Microbiome and Intestinal Pathology in Parkinson’s Disease
Article information

Abstract
Parkinson’s disease (PD) is a common neurodegenerative disorder arising from an interplay between genetic and environmental risk factors. Studies have suggested that the pathological hallmarks of intraneuronal α-synuclein aggregations may start from the olfactory bulb and the enteric nervous system of the gut and later propagate to the brain via the olfactory tract and the vagus nerve. This hypothesis correlates well with clinical symptoms, such as constipation, that may develop up to 20 years before the onset of PD motor symptoms. Recent interest in the gut–brain axis has led to vigorous research into the gastrointestinal pathology and gut microbiota changes in patients with PD. In this review, we provide current clinical and pathological evidence of gut involvement in PD by summarizing the changes in gut microbiota composition and gut inflammation associated with its pathogenesis.
Parkinson’s disease (PD) is a common neurodegenerative disorder arising from the interplay between genetic and environmental factors. In addition to the well-known motor symptoms of bradykinesia, rigidity, rest tremor, and postural instability, PD also involves various nonmotor symptoms, including constipation, depression, sleep disturbance, and hyposmia. Among these, constipation is the most common and can precede motor symptom onset by more than a decade [1]. Intraneuronal α-synuclein aggregations, the pathological hallmarks of PD, were recently identified in the olfactory bulb and enteric nervous system (ENS) in patients with PD based on postmortem pathological examinations [2-4]. Given the topographical distribution of intraneuronal α-synuclein deposits, known as Lewy bodies (LBs), Braak and coworkers hypothesized that PD pathology may involve transsynaptic cell-to-cell transmission of α-synuclein from the ENS via the vagus nerve and the olfactory bulb to the substantia nigra and further areas of the brain [5]. Combined with the fact that the ENS and olfactory bulb are the gateways to the environment, this evidence suggests an involvement of environmental factors in PD pathogenesis [6].
Findings from several subsequent studies are in line with Braak’s hypothesis, although there are some conflicting results. Bowel inflammation triggered by rotenone, immune activation by Escherichia coli (E. coli)-producing amyloid protein curli or bacterial products (lipopolysaccharide, LPS) induce α-synuclein aggregations in the gut or both in the gut and brain [7-9]. Animal studies have shown that α-synuclein can spread from the gut to the brain via the vagus nerve [10,11] and that vagus nerve resection can successfully stop this transmission [9]. Recent findings point to consistent gut microbiota changes in patients with PD compared to ageand sex-matched healthy controls [12]. However, despite a plethora of evidence suggesting a role of the gut–brain axis in the development of PD, some conflicting results exist. As many as 47% of PD cases from a UK tissue bank and 18% from a Vienna brain tissue bank did not show the pattern of pathological α-synuclein distribution proposed by Braak and colleagues [13,14]. In addition, the results of epidemiological studies have been inconsistent about the risk reduction of PD in patients who have undergone vagotomy [15-17].
In this review, we provide the current clinical and pathological evidence of gut involvement in patients with PD and the possible role of gut microbiota changes as potential environmental triggers in the disease process.
GASTROINTESTINAL SYMPTOMS IN PATIENTS WITH PD
Gastrointestinal (GI) impairments are commonly observed at all stages of PD, with almost 30% of patients reporting GI symptoms, including drooling, dysphagia, gastroparesis, and constipation [18]. The prevalence of drooling in PD ranges from 10% to 84% [18-22]. The presentation is related to swallowing dysfunction in the oropharyngeal phase [19] and an increased rate of parotid gland secretion [20] that worsens with a flexed posture, unintended mouth opening, and divided attention [21]. The prevalence of dysphagia ranges from 9% to 82% but has been up to 97% in objective studies [22]. Dysphagia usually develops in patients with advanced PD who have severe bradykinesia and rigidity, which is thought to contribute to oropharyngeal dysphagia [23]. The prevalence of gastroparesis ranges from 70% to 100%, with an average half-emptying time of 46 to 149 minutes in patients with mild PD and 55 to 221 minutes in moderate/severe PD, compared to 43 to 107 minutes in healthy controls [24]. Although the exact pathophysiology is unclear, gastroparesis is a major player in the development of motor fluctuations in PD [25].
The most common GI symptom in patients with PD is constipation, which is also among the most prevalent prodromal symptoms of PD [26]. The prevalence of constipation in PD ranges from 8% to 70% and increases with disease progression [27]. Evidence suggests that this prevalence is up to 6‑fold greater among patients with PD than among age-matched and sex-matched controls [28]. An increasing number of studies have evaluated the temporal relationship between bowel movement frequency and PD risk. The Honolulu-Asia Aging cohort study in males, which had a 24-year follow-up period, showed that infrequent bowel movements, as assessed by a self-reported bowel habit questionnaire, were associated with an increased risk of PD and reduced neuronal density in the substantia nigra [29]. Our study using a population-based national database also showed that the severity of constipation, quantitatively defined by the amount of laxative use, was dose-dependently associated with future PD risk, independent of age, sex, underlying comorbidities, or medication use [30]. A case–control study reported a greater PD risk among individuals with a history of constipation, as early as 20 years before the onset of motor symptoms, as assessed by a review of medical records [31]. PD-related constipation may be traced to LB deposition in the ENS or the dorsal motor nucleus of the vagus nerve, which are among the earliest affected regions in PD [32]. Despite vigorous research, the precise etiology of constipation and whether constipation in PD is caused by gut or brain pathology remain elusive [33].
GASTROINTESTINAL PATHOLOGY FINDINGS IN PATIENTS WITH PD
The GI tract consists of four layers: the mucosa, submucosa, muscular layer, and serosa (ordered from innermost to outermost). It contains its own intrinsic nervous system, the ENS, but is also modulated extrinsically by the autonomic nervous system, which is involved in PD [34]. The ENS consists of submucosal plexuses and myenteric (Auerbach) plexuses (Figure 1). The submucosal plexuses have three layers: the inner (Meissner plexus), intermediate, and outer (Schabadasch or Henle) plexuses, which are mainly responsible for secretion and blood flow in the GI tract. Myenteric plexuses mostly control intestinal peristalsis [35]. Virtually every neurotransmitter produced by neurons in the central nervous system (CNS) have also been identified within the ENS, including acetylcholine, nitric oxide (NO), vasoactive intestinal peptide (VIP), and catecholamines [36]. Autonomic parasympathetic input mediates the central modulation of ENS function primarily from the dorsal motor nucleus of the vagus nerve and the sympathetic input from the para- and prevertebral ganglia.

Vagal parasympathetic nerve fibers connecting the ENS and CNS. ENS: enteric nervous system, CNS: central nervous system, Ach: acetylcholine, VIP: vasoactive intestinal peptide.
The accumulation of α-synuclein is not limited to central dopaminergic neurons, and a growing body of literature has shown that α-synuclein aggregation can be detected particularly in the ENS at postmortem and in safely accessible in vivo biopsies of the GI tract. Some studies have even demonstrated detectable α-synuclein pathology in the GI tract in the prodromal phase of PD. Below, we summarize the recent GI pathology findings in patients with PD according to the findings from postmortem pathology studies, colon biopsy (only covering the mucosal and submucosal layers) and surgical colectomy findings (covering all three layers of colon) of PD patients.
Evidence from postmortem autopsy studies
In 1984, Qualman and colleagues [37] first reported the possible presence of LBs in the GI tract, based on the autopsies of 8 patients with achalasia, 22 with PD, and 50 controls. Using hematoxylin-eosin (H&E) staining, they examined multiple levels of the entire GI tract (esophagus, stomach, jejunum, ileum colon, and rectum). LB-like intraneuronal inclusions were found in the myenteric plexus of the esophagus in 2 of 8 patients with achalasia and 1 of 3 PD patients with dysphagia, but none were found in the controls. This intraneuronal inclusion pathology was most prominent in the distal esophagus in the achalasia patient who had the shortest duration of the disease and retained degenerated ganglion cells in the esophagus. The inclusions were also found in the myenteric plexus but not the submucosal plexus of the colon in one PD patient who had dysphagia. Subsequent autopsy reports by Wakabayashi and colleagues [38,39] using H&E staining and electron microscopy described LB-like inclusions in both the myenteric plexus and submucosal plexus and throughout the GI tract in PD patients, more numerously in the distal esophagus. Intriguingly, small LBs were also observed in the myenteric plexus of the esophagus or small intestine in 8 of 24 age-matched controls who did not have PD [39]. Further immunohistochemistry staining of different types of neurons of 3 autopsied PD patients showed that the LBs resided in the VIP neurons instead of tyrosine hydroxylase neurons in the paravertebral and celiac sympathetic ganglia and the ENS of the GI tract [38].
In 2006, Braak and colleagues [40] investigated the gastric pathology of 5 patients with pathologically confirmed PD using immunohistochemistry with an antibody against α-synuclein (anti-Syn-1, clone 42). α-synuclein pathology, in the form of LBs and immunoreactive fibers, was identified in the myenteric plexus, submucosal plexus, and peripheral nerves in the adventitia of the distal esophagus and stomach in all PD patients (5/5) but in no controls (0/5) (Table 1). One of the five PD patients had α-synuclein pathology in the ENS and dorsal motor nucleus of the vagal nerve without α-synuclein deposits in the substantia nigra. These findings, in combination with the same group’s discovery of an α-synuclein staging pattern in the CNS [32], led to Braak and colleagues’ pathology hypothesis of PD [3,4] and opened the door to subsequent vigorous research on α-synuclein in the gut.

Summary of staining results using immunohistochemistry methods targeting α-synuclein in GI tracts, from autopsy reports
The autopsy reports of immunohistochemistry staining targeting α-synuclein in the GI tracts are summarized in Table 1. Most studies used antibodies against α-synuclein, although with different clones. The frequency of α-synuclein detection in PD patients was higher than in controls in most studies (Table 1) [40-45]. One group using an antibody targeting phosphorylated Ser 129 α-synuclein obtained positive results in 11 of 17 patients with PD and in 0 of 23 controls [46]. In addition to patients with PD, α-synuclein pathology was observed in patients with diffuse LB dementia (DLB), comprising 5 of 9 patients with DLB in one study and 5 of 5 in another; on the other hand, it was rarely found in patients with Alzheimer’s disease [44,45] and not found in 2 patients with multiple system atrophy in a study that examined only the esophagus [43].
These autopsy reports using immunohistochemistry to target intraneuronal α-synuclein were in agreement with a rostro–caudal gradient pattern of α-synuclein distribution in the GI tract, with more abundant α-synuclein pathology in the esophagus and stomach compared to the colon [41,44,46]. In addition, α-synuclein pathology was found in both the myenteric and submucosal plexuses but more frequently in the former. LBs, if present, were mostly found in the myenteric plexus. Notably, Annerino and colleagues [41] reported that in contrast to earlier findings of Wakabayashi and colleagues, α-synuclein pathology did not colocalize with NO-positive or VIP-positive neurons and did so with less than 3% of the tyrosine hydroxylase–positive neurons. PD motor symptom severity also did not correlate with the density of enteric neurons or α-synuclein pathology in that study [41].
Evidence from colon biopsy studies
Following the initial autopsy reports of α-synuclein in the GI tract [40], many studies addressed the feasibility of detecting α-synuclein as a peripheral biomarker using colonic biopsy specimens of living patients. The results of the biopsy reports are summarized in Table 2. Overall, they were inconsistent. Because of safety concerns, the specimens from a biopsy procedure contain only the mucosa and, at most, the submucosa. Based on immunohistochemistry, the frequency of α-synuclein or phospho-α-synuclein in the stomach mucosa ranged from 31.6% to 100% in PD patients and 4.3% to 41.7% in controls [47-49]; the frequency of α-synuclein or phospho-α-synuclein in the mucosal layer of the colon was 0% to 100% in PD patients and in controls [47,48,50-53]; and the frequency of α-synuclein or phospho-α-synuclein in the submucosal layer of the colon was 5% to 100% in PD patients and 0% to 100% in controls [52-59]. Some groups examined α-synuclein or phospho-α-synuclein in various regions of the mucosa or mucosal layers of the GI tract and found frequencies from 9% to 100% in PD patients and 0% to 36% in controls [60-64]. Although the findings of many studies still supported a higher frequency of α-synuclein or phospho-α-synuclein in PD patients than in controls [49,53-59,61,62], others suggested similar frequencies between groups [47,48,50-52,60,63-65].

Summary of staining results using immunohistochemistry methods targeting α-synuclein in gastrointestinal tracts, from biopsy reports
Despite the higher frequency of α-synuclein or phospho-α-synuclein deposits in colonic biopsy specimens in PD patients versus controls in some studies, others offered conflicting results (Table 2). Some explanations for the discrepancies include the variable individual GI conditions of participants, different PD subtypes and disease durations, variable location and depth of the biopsies, the number of biopsies and slides examined, specimen size, different definitions for α-synuclein positivity, different neuronal markers for nerves and neurons, different antibodies, and most important, different specimen preparation (e.g., whole-mount with microdissection vs. formalin-fixed, paraffin-embedded) and immunohistochemistry methods.
Although some studies uncovered a rostro–caudal gradient of α-synuclein deposits in colonic biopsies [47,57,62], other studies did not [52,61]. α-synuclein pathology can also be found in the preclinical stage of PD patients [58,61] and in patients with idiopathic REM sleep behavior disorder [52]. The proportion of α-synuclein positivity did not differ between patients with preclinical PD and those with motor symptoms of PD in one study (10.6% were in the preclinical stage, defined as up to 8 years prior to clinical diagnosis; 10.7% had PD motor symptoms) [61]. The difference between preclinical PD and controls was detected only with an antibody against phospho-α-synuclein in another study (phospho-α-synuclein positivity in PD vs. preclinical PD vs. controls: 48%, 45%, and 26%, respectively; total α-synuclein positivity in PD vs. preclinical PD vs. controls: 61%, 52%, and 47%, respectively) [65]. Additionally, biopsy positivity did not differ between patients in the early and late stages of PD [51].
Few studies have evaluated the correlation between motor symptom severity and α-synuclein pathology of the GI tract. Lebouvier and colleagues [56] reported a positive correlation of axial symptom subscores of the unified PD rating scale (UPDRS), levodopa responsiveness, and chronic constipation severity with LB pathology in the submucosa of the GI tract. Hilton and colleagues [61] found that all PD patients with positive findings of α-synuclein pathology in colonic biopsies had early autonomic symptoms, although other findings conflicted with these results [49,47,63].
Evidence from surgical specimens of colectomy
Surgical specimens are superior to biopsy specimens because they allow for better specimen quality with adequate tissue and full thickness of the intestinal wall. However, the disadvantage of studies using surgical specimens is that only diseased specimens can be obtained, which can affect the results. The results of reports based on surgical specimens are summarized in Table 3. Shin and colleagues [64] compared phospho-α-synuclein positivity (EP1536Y) in colonic biopsy specimens with surgical specimens from patients with PD. Of interest, although the stomach surgical specimens had a higher positive rate for phospho-α-synuclein immunoreactivity in PD participants (58%) than in controls (8.3%), the colorectal surgical specimens did not differ (PD patients vs. controls: 23.8% vs. 23.8%). Furthermore, the positivity of colorectal mucosal biopsy specimens did not differ between PD patients (9.1%) and controls (18.2%) [64]. In comparison studies between PD patients and controls, the overall frequency of α-synuclein pathology in PD patients detected using surgical specimens has been 36% to 100%, higher than in the controls (0% to 22%) [64,66-68].
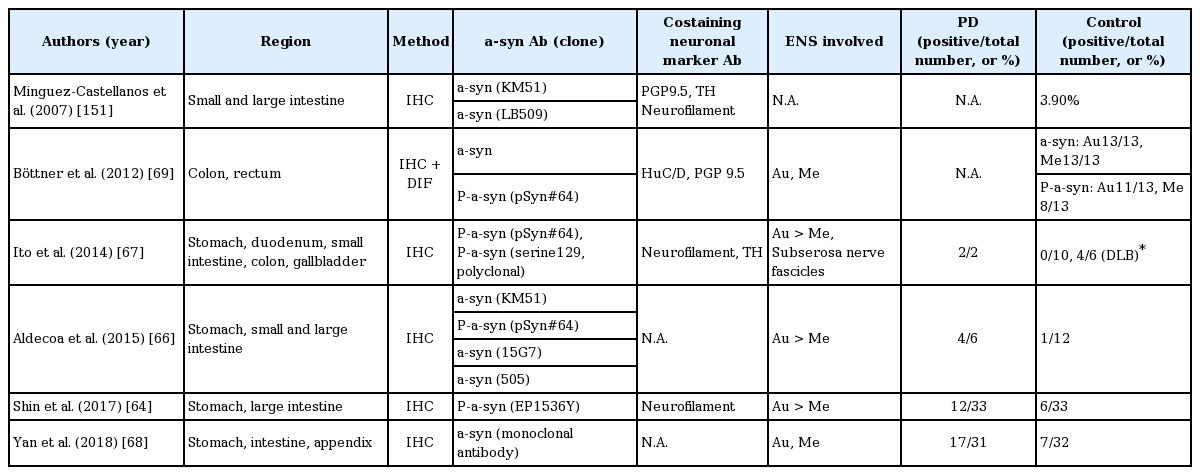
Summary of staining results using ICH methods targeting α-synuclein in gastrointestinal tracts, from surgical specimens
Yan and colleagues [68] studied surgical specimens, including colon and stomach, using an α-synuclein mouse monoclonal antibody and found that α-synuclein pathology is most frequent in the body of the stomach in PD patients but not in controls. Using different primary antibodies, Aldecoa and colleagues [66] detected LB-like intraneuronal aggregations in the body of the stomach in 4 out of 6 PD patients but in only 1 out of 12 controls. Additionally, the pattern of punctate cytoplasmic staining of ganglion cells was observed only in staining with anti-phosphorylated Ser129 α-synuclein antibody [66]. However, some studies have produced conflicting results, identifying α-synuclein pathology in the submucosal and muscular layers of the colon or appendiceal mucosal nerve plexus in neurologically healthy controls who underwent partial colectomy or sigmoid resection [69,70].
These conflicting results have led to several investigations into the sensitivity and specificity of different conventional immunohistochemistry methods or to attempts with other methods to produce more consistent results. Corbillé and colleagues [71] distributed a common set of colon biopsy slides from 9 PD patients and 3 controls to four different laboratories for immunohistochemistry staining using different α-synuclein or phospho-α-synuclein antibodies and epitope exposure methods. Their results showed that no particular single staining method or staining pattern had a sensitivity and specificity of more than 80%. The same group further examined full-thickness sigmoid colon specimens from 5 pathology-confirmed PD patients and 5 controls using different immunohistochemistry methods and antibodies [anti-phospho-Ser129 α-synuclein, including EP1536Y (ab51253) and MJF-R13 (8-8) (ab168381), anti-phospho-Ala81 α-synuclein, polyclonal p-synuclein LB509], and epitope exposure methods [72]. They obtained excellent accuracy with 100% specificity and sensitivity most frequently in the submucosa instead of the muscularis layer with the combination of a specific staining pattern (“fibers and puncta” and “fibers only” representing neuronal distribution), a particular method with primary antibody targeting anti-phospho-Ser129 α-synuclein {[MFJ-R13 (8-8)] ab168381 with a concentration of 1:5,000, and an epitope exposure method using proteinase K with a concentration of 1:100 in 37°C for 20 minutes}, and raters with good interrater agreement [72]. Therefore, an immunohistochemistry study for assessing α-synuclein pathology in the colon is reliable only if adequate submucosa tissue is obtained, a larger amount of α- synuclein pathology is present, a specific staining pattern associated with neuronal morphology is recognized, a suitable immunohistochemistry method is used, and raters are well trained [72].
Punsoni and colleagues [73] performed quantitative polymerase chain reaction (qPCR) in addition to conventional immunohistochemistry methods in 4 groups of participants, including living PD patients, PD autopsy cases, living pediatric controls, and living adult controls. The specimens included different regions of the GI tract and of different thicknesses, including submucosa and myenteric plexuses. The expression of α-synuclein in neurons was confirmed by colocalization with neurofilament staining. Immunohistochemistry analysis showed that although α-synuclein staining was diffusely present in all PD patients and controls, LBs were observed in the myenteric plexus of 25% of PD patients but not in controls. Moreover, the mean α-synuclein expression examined by qPCR was highest in PD patients and lowest in controls [73]. Visanji and colleagues [51] used proteinase K to enhance antigen retrieval in immunohistochemistry analysis in paraffin-embedded tissues and found a lower positive rate of either total or phospho-α-synuclein immunoreactivity in PD patients than in controls. Barrenschee and colleagues [60] used morphometric analysis followed by dual-label immunohistochemistry and found that the total numbers and areas of phospho-α-synuclein aggregates per neuron were increased in PD patients compared with controls.
Summary of gastrointestinal pathology findings in patients with PD
In summary, no consensus technique has yet been established for detecting α-synuclein pathology in gut tissues that can reliably differentiate PD patients from controls. To allow clinicians to use α-synuclein pathology staining in the GI tract as a biomarker for PD pathogenesis, the development or testing of other techniques is warranted. This process should include both quantitative and qualitative methods in addition to careful stratification of PD patients into different subtypes, focusing on those who are most likely to be in line with Braak and colleagues’ hypothesis.
MECHANISM OF GI IMPAIRMENT AND GUT PATHOLOGY IN PD
Neuronal loss in the ENS in PD
In the first autopsy report of LBs in the ENS using H&E staining, degeneration of enteric ganglion cells was found in the myenteric plexus in the esophagus of one patient with PD who had dysphagia [37]. A subsequent autopsy report of one patient with PD and acquired megacolon found well-preserved ganglia and ganglion cells in both the submucosal and myenteric plexuses of the colon using H&E staining [74]. Neuron counting via immunohistochemistry or coimmunofluorescence staining also showed variable results in both the submucosa and the muscular layer. Lebouvier and colleagues [55,75] examined colonic biopsy specimens with the mouse pan-neuronal marker anti-Hu C/D and found no difference between PD patients and controls in the neuron number per ganglion or proportion of dopaminergic neurons in the submucosal layer. However, their subsequent study showed a decreased number of neurofilament heavy chainimmunoreactive neurons per ganglion in patients with positive phosphorylated α-synuclein aggregations in the submucosa. Furthermore, the LB pathology burden was negatively correlated with neuronal count and levodopa responsiveness but positively correlated with axial symptoms and constipation severity [56]. On the other hand, a recent study examining rectal biopsy specimens using the pan-neuronal marker PGP9.5 showed that patients and controls had similar mean neuronal area, ganglionic area, and neuron number per ganglion in the submucosa of rectum [60]. The divergent findings in these studies are probably the result of the randomly biopsied regions and different methods used. Therefore, although most patients with PD have constipation, it may not be directly related to neuronal loss in the ENS; instead, the extrinsic autonomic system probably plays a role [34].
Evidence of enteric glial cells involved in PD
Enteric glial cells (EGCs) are another population of cells in the ENS known for a role in neuroprotection, gut inflammation, intestinal epithelial barrier function, and synaptic neurotransmission regulation [76]. As there was no overt neuronal loss identified in the ENS, another possible cause of GI dysfunction may be traced to the EGCs. Despite a large variation among patients, two studies suggested that reactive gliocytosis (severe glia reactions) might be present in the bowel given the general increase in the expression of the glial markers, glial fibrillary acidic protein and Sox-10 [77,78]. The underlying mechanism of these phenomena remains elusive, and further studies are warranted to elucidate the involvement of EGCs in the PD process.
Evidence of leaky gut and bowel inflammation in PD
Recent animal studies have shown that bowel inflammation can exacerbate neuroinflammation, disrupt the blood–brain barrier, and promote dopaminergic neuronal loss in the substantia nigra of an inflammation rat model of PD. In this model, substantia nigra inflammation and selective dopaminergic neuronal loss were induced by LPS injection [79]. In agreement, several studies have shown clinical evidence of bowel inflammation in PD patients. Devos and colleagues [78] used real-time PCR to analyze the mRNA expression of pro-inflammatory cytokines and glial markers in the ascending colon biopsies of PD patients and controls. They found significantly increased expression of proinflammatory cytokines [tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin (IL)-6, and IL-1β] in PD patients. This increase correlated with increased expression of glial markers (glial fibrillary acidic protein and Sox-10), indicating that pro-inflammatory processes in the bowel are enhanced in PD patients. Of note, the authors found that expression levels of cytokines and glial markers did not correlate with immunostaining levels of phospho-α-synuclein and axial symptom subscores on the UPDRS part III. In addition, these expression levels correlated negatively with PD duration. This observation suggests that bowel inflammation may play a role in PD pathogenesis [78].
Consistent with this possibility, stool immune profiles show elevations in proteins related to angiogenesis and in chemokines and cytokines such as IL-1α, IL-1β, and IL-8 in PD patients compared to controls [80]. Another study also reported increases in the fecal intestinal permeability marker fecal calprotectin in patients with PD compared to age-matched controls; however, the level of fecal calprotectin did not correlate with any clinical parameters, including disease duration, in patients with PD [81].
Recent evidence suggests that gut inflammation in PD may be traced to increased intestinal permeability, also known as the leaky gut. This status correlates with intestinal α-synuclein accumulation in patients with PD [82]. Increased intestinal permeability and translocation of bacteria and inflammatory bacterial products (e.g., LPS) might lead to inflammation in the GI tract and thereby initiate α-synuclein accumulation in the ENS [82]. Forsyth and colleagues [82] also observed increased urine sucralose excretion in patients with PD compared to controls, suggesting increased colon permeability. Furthermore, increased colon permeability correlates with increased α-synuclein accumulation pathology and E. coli immunohistochemistry staining of distal sigmoid biopsy in PD patients [83]. This observation supports the hypothesis that increased permeability-related bowel inflammation with an increased chance of gut bacteria translocation may be involved in PD pathogenesis.
Clairembault and colleagues [54] further examined morphological changes and expression in the intestinal epithelial barrier (including two tight junction proteins, ZO-1 and occludin) in colonic biopsy tissues of PD patients and controls. They found that a larger proportion of PD patients (14 of 31 patients) had disrupted and irregularly distributed tight junction proteins and lower expression levels of occludin compared with controls (1 of 8 controls) [54]. These observations provide some evidence of increased gut permeability and mild bowel inflammatory changes observed in patients with PD.
The link between bowel inflammation and PD also comes from recent findings of common genetic risk variants between inflammatory bowel disease (IBD) and PD [84]. Among these candidates are two genes, NOD2 and LRRK2, the latter of which is the most common causative genetic variant linked to PD. They are of special interest because of their involvement in the innate immune system, which regulates the immune response [85]. Genetic findings have led to several epidemiological studies examining the relationship of PD risk in patients with IBD. Four large population studies from Taiwan (person-years: 299,900) [86], Denmark (person-years: > 8,000,000) [87], the United States (personyears: 3,008,825) [88], and Sweden (person-years: 249,784) [89] have demonstrated increased PD risk in patients with IBD. PD incidence thereby increased by 35, 22, 28, and 30% in patients with IBD in the Taiwanese, Danish, American, and Swedish populations, respectively [86-89]. The risk was higher in patients with Crohn’s disease (CD) than in those with ulcerative colitis (UC) in the Taiwanese population but higher in those with UC than in those with CD in the Swedish and Danish studies [87,89]. In the US study, the PD risk increase was similar for CD and UC [86-89].
Consistently, two US studies also showed that treatment of IBD, one with anti-TNF therapy [88] and the other with unspecified immunosuppressants [90], reduced PD risk. In the Swedish study, patients with IBD who did not receive treatment were 60% more likely to develop PD than their matched controls, whereas treated IBD patients had a nonsignificantly decreased risk of developing PD compared to nontreated patients [89]. However, a retrospective study enrolling 876 patients with PD identified only 2 patients with CD prior to the PD diagnosis, an incidence similar to that in the general population [91]. Another large population-based, case–control study enrolling patients aged 65 years or older with newly diagnosed PD (n = 89,790) and controls (n = 118,095) identified an inverse correlation between PD and IBD, suggesting that patients with IBD are less likely to develop PD [90].
Despite the inconsistent results, these recent pathological, genetic, and epidemiological studies have suggested a relationship between bowel inflammation and PD. The exact mechanism linking bowel inflammation and neurodegeneration in the disease process of PD is still elusive and requires further studies in the future, which would shed light on potential treatment strategies for halting the neurodegenerative process of PD.
ALTERED GUT MICROBIOME IN PATIENTS WITH PD
Gastric Helicobacter pylori and small intestinal microbial overgrowth
Gastric Helicobacter pylori infection is a common chronic infection that has been implicated in PD [92-101]. Most epidemiological studies using different detection methods (ELISA, urea breath test, RT-PCR, histological examination) have shown an increased risk of H. pylori infection in PD, ranging from 1.3- to 2-fold (prevalence: 22% to 70%) [92-101], which was also shown to be significant in a recently published meta-analysis [102]. Meta-analyses have also identified a significantly worse mean UPDRS score in PD patients with H. pylori infection (either UPDRS or total UPDRS III in “on” or “off” state) [97,98,103-107] and improvement in UPDRS part III scores after H. pylori eradication [102-105,108,109]. The exact mechanisms of involvement of chronic H. pylori infection in PD pathogenesis are unclear but possibly trace to multifactorial inputs, including the H. pylori toxin, neuroinflammation, and alterations in the gut microbiome [110].
Small intestinal bacterial overgrowth (SIBO) is also more prevalent in patients with PD than in healthy controls in crosssectional studies, with a prevalence of 25% to 54% [97,111-114]. Most studies used lactulose or glucose breath tests to detect SIBO, with sensitivities of 31–68% and 29–93%, respectively, and specificities of 44–100% and 30–86%, respectively [115]. Bloating and flatulence [112,113] have been reported to be associated with SIBO in PD in some studies, whereas constipation and tenesmus were reported in one study [114]. The relationship between PD symptoms and SIBO is heterogeneous among reports. Disease duration, Hoehn and Yahr stage, UPDRS, motor scores, motor fluctuations, or nonmotor symptom severity have all been mentioned in association with SIBO [97,112-114]. Treatment of SIBO is reported to have improved motor fluctuation symptoms in PD patients, but the recurrence rate at 6 months was as high as 43% [97].
Altered gut microbiota in PD
To date, 13 studies (including one published abstract) have compared the difference in gut microbiota composition between PD patients and controls (Table 4 and 5) [116-128]. While α-diversity (species diversity within a single subject) was similar in most studies [116,120,122,123,126,127], two showed increased [121,125] and two showed decreased α-diversity [124]. All studies revealed differences in microbiota composition between PD patients and controls (β-diversity), but the results were heterogeneous (Table 4). In brief, when compared with controls, the family Verrucomicrobiaceae (phylum Verrucomicrobia) and genera Akkermansia (phylum Verrucomicrobia; family Verrucomicrobiaceae) and Lactobacillus (phylum Firmicutes; family Lactobacillaceae) were increased in PD patients in several studies [116,118,119,126]. On the other hand, the families Prevotellaceae (phylum Bacteroidetes), Lachnospiraceae (phylum Firmicutes), and Pasteurellaceae (phylum proteobacteria) and genera Blautia (phylum Firmicutes; family Lachnospiraceae), Roseburia (phylum Firmicutes; family Lachnospiraceae), Prevotella (phylum Bacteroidetes; family Prevotellaceae), and Faecalibacterium (phylum Firmicutes; family Clostridiaceae) were decreased in PD patients in three or more studies [116,126,128]. Prevotella is abundant in humans that consume plant-based, fiber-rich diets, which are substrates for bacteria to produce short chain fatty acids (SCFAs) [129]. Prevotella and Akkermansia are both mucin-degraders that promote and serve as indicators of gut integrity [130]; however, they both have also been linked to gut or systemic inflammatory conditions [129,131]. On the other hand, Faecalibacterium prausnitzii has anti-inflammatory properties other than an effect of butyrate production [132-134]. Of interest, similar to PD patients, Faecalibacterium prausnitzii and Lachnospiraceae are depleted in patients with IBDs. In contrast to PD patients, however, Akkermansia is decreased in patients with IBDs [135].
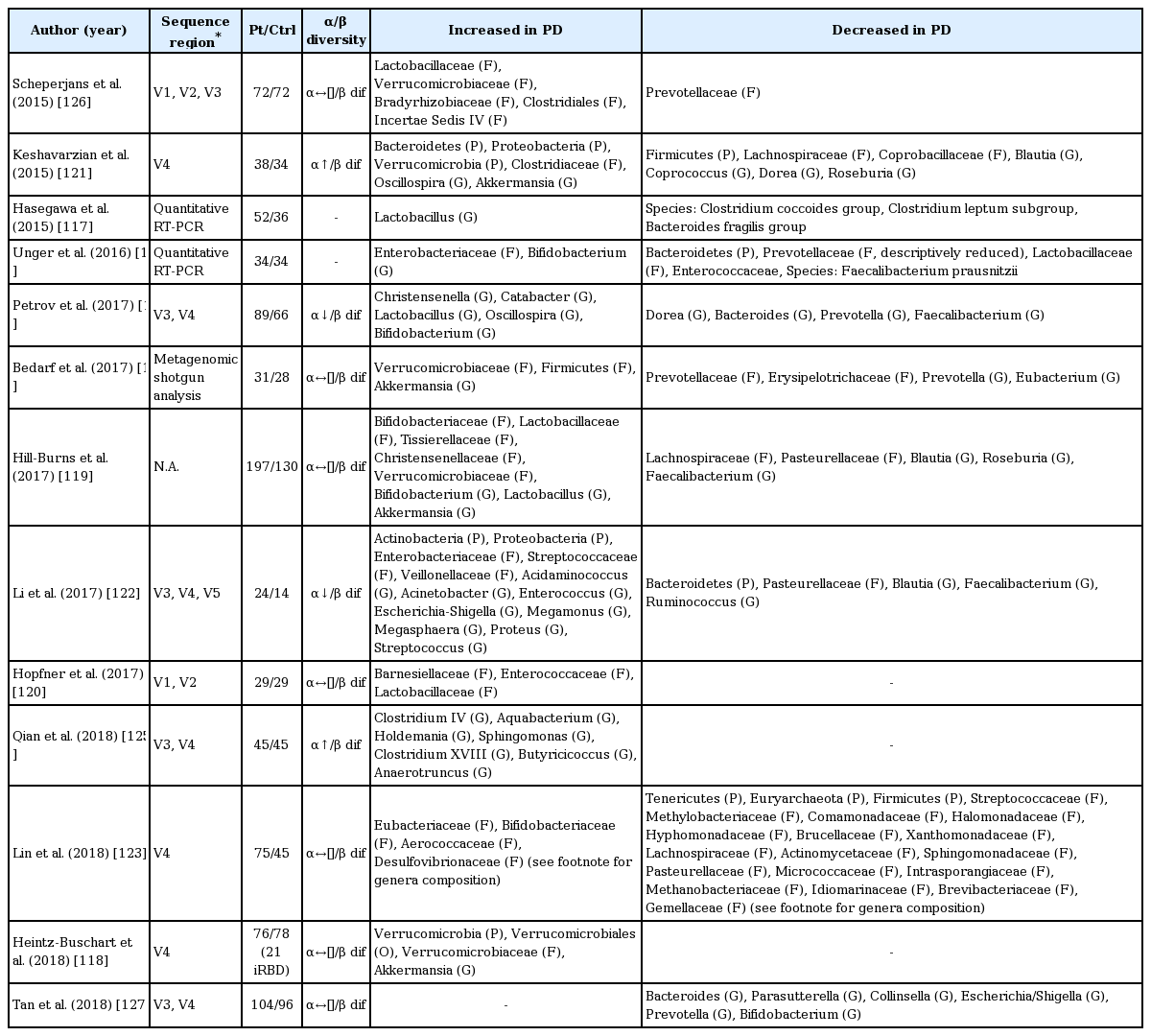
Summary of altered microbiota compositions in patients with PD compared with healthy controls

Differences in bacterial taxa between PD patients and controls detected in more than two studies
Some groups have tried to delineate the relationship between fecal microbiota and clinical features but with heterogeneous results. Two studies identified a correlation of PD duration with the abundance of the genera Escherichia/Shigella or family Enterobacteriaceae among other nonoverlapping bacteria [122,124]. The abundances of Enterobacteriaceae also correlated with the severity of postural instability and gait disturbance in another independent study [126].
Altered gut metabolites in PD
The metabolites of gut microbiota, SCFAs, are produced by nondigestible carbohydrate fermentation via different metabolic pathways. The three most abundant SCFAs in the human colon are acetic acid, propionic acid, and butyric acid [136]. Different SCFA-producing bacteria adopt different metabolic pathways using different substrates and enzymes to selectively produce different SCFAs [136]. Many of these altered bacteria, e.g., from the families Prevotellaceae and Lachnospiraceae and genera Akkermansia, Blautia, Roseburia, and Faecalibacterium, comprise species that are SCFA producers and are decreased in PD patients except for the genus Akkermansia. One study also identified reduced fecal SCFA concentrations in PD patients [128].
Accumulating evidence has suggested a role for SCFAs in maintaining gut barrier function and regulating gut motility and the immune response in the gut. One of the mechanisms for SCFAs in regulating the immune system in the gut is through gene expression control via histone deacetylase inhibition. This inhibition in colonocytes and immune cells by butyrate and propionate results in an increased expression of the anti-microbial cathelicidin-derived peptide of the innate immune system, IL-37, anti-inflammatory cytokines, IL-10, and transforming growth factor β [137]. It is also associated with the downregulation of proinflammatory cytokines, including IL-8, IL-6, IL-1β, IFN-γ, and TNF-α, and the promotion of colonic regulatory T cell differentiation to control intestinal inflammation [138]. Furthermore, butyrate, propionate, and acetate can interact with different G protein-coupled receptors (GPRs), including GPR41 (FFA3), GPR43 (FFA2), and GPR109A, located on the surface of host cells. There, they also exert an anti-inflammatory effect of T cell differentiation and modulation and NF-κB activation inhibition [138,139]. Despite evidence suggesting an intestinal health benefit of SCFAs, the major controversy for the role of gut microbiota in PD comes from a recent study showing that microbiota and SCFAs in the gut exacerbate α-synuclein pathology and microglia cell activation in α-synuclein–overexpressing mice [140]. Another detrimental effect of SCFAs, in particular propionic acid, was also shown to be related to other neurological disorders, such as autism, although the underlying mechanism remains unknown [141]. Different SCFAs have different effects on biological systems. To further delineate the role of SCFAs in PD, future studies should evaluate the individual SCFAs and their balance with each other instead of focusing on their effects as a whole.
More research is needed to determine not only bacterial composition in PD patients but also changes in related biochemical pathways. These studies can rely on methods such as metabolomics, proteomics, metatranscriptomics, and shotgun metagenomic sequencing, which could lead to further insights into the role of altered gut microbiota and metabolites in the pathogenesis of PD.
The effects of anti-PD medications on the gut microbiota
One recent study found a significant difference in the gut microbiome as a function of treatment with COMT inhibitors and anticholinergics, and a borderline significance for carbidopa/levodopa [119]. As the growing literature on the role of the gut microbiome in the metabolism of medications and the profound effects that the drugs can have in turn on the composition of the microbiome [142-145], the interaction between use of PD medications and the compositions of gut microbiome is not surprising. Another prior study of PD also linked COMT inhibitors to altered abundance of some taxa [126]. Additionally, COMT inhibitors and anticholinergics have gastrointestinal side effects, which may contribute to altered gut microbiome [146,147]. These findings lend support to the notion that there is a clinically important relationship between microbiota and drug metabolism throughout the lifespan of PD patients; therefore, profiling of the human microbiome will be essential to understand the mechanisms by which these microbiota–drug interactions occur and the degree to which this complex interplay affects drug efficacy. Additional studies are needed to assess the potential utility of bacterial therapeutics in altering the microbiome to enhance therapeutic efficacy and clinical outcomes in patients with PD.
IMPLICATIONS FOR FUTURE GUT-TARGETED THERAPY FOR EARLY-STAGE PD
Recent advances in understanding the gut–brain axis have linked microbiota and intestinal pathology to PD pathogenesis (see review [148]). Based on animal and clinical studies, changes in the composition of the gut microbiota are proposed to induce a pro-inflammatory response in the gut. According to this conceptualization, the inflammatory response increases gut permeability and exposure to endotoxins or other bacterial products and induces α-synuclein aggregations, which in turn propagate to the CNS via the vagus nerve. In addition, a systemic inflammatory response might disrupt the blood–brain barrier, which, in combination with α-synuclein delivery from the gut, further activates microglia and results in dopamine neuron degeneration (Figure 2) [12]. However, many pieces remain missing from this huge puzzle of the gut–brain axis in PD. Future investigations are necessary to define specific microbe-derived factors, immune effector functions, and microbiota–immune pathways for modulating gut–brain communications. These studies will reveal fundamental biological insights into PD pathogenesis, with the potential to inform the development of new microbial- and immune-based therapeutic strategies for treatment.

Vagal and nonvagal pathways involved in bidirectional communication between the gut microbiota and the brain. SCFAs: short chain fatty acids.
Notes
Conflict of interest
The authors have no financial conflicts of interest.
Acknowledgements
The authors are grateful to the National Health Research Institutes (NHRI-EX107-10716NC) and the National Taiwan University Hospital (106-EDN06 and 107-EDN14) for their funding support of this work.