E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(2); 2018 > Article
-
Case Report
A Patient with Beta-Propeller Protein-Associated Neurodegeneration: Treatment with Iron Chelation Therapy -
Shen-Yang Lim1,2
 , Ai Huey Tan1,2, Azlina Ahmad-Annuar3, Susanne A. Schneider4, Ping Chong Bee5, Jia Lun Lim2,3, Norlisah Ramli6, Mohamad Imran Idris1
, Ai Huey Tan1,2, Azlina Ahmad-Annuar3, Susanne A. Schneider4, Ping Chong Bee5, Jia Lun Lim2,3, Norlisah Ramli6, Mohamad Imran Idris1 -
Journal of Movement Disorders 2018;11(2):89-92.
DOI: https://doi.org/10.14802/jmd.17082
Published online: May 30, 2018
1Divisions of Neurology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
2The Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, University of Malaya, Kuala Lumpur, Malaysia
3Department of Biomedical Science, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
4Department of Neurology, University of Munich, Munich, Germany
5Divisions of Haematology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
6Divisions of Neuroradiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- Corresponding author: Shen-Yang Lim, MBBS, MD, FRACP, FASc, https://orcid.org/0000-0002-6942-2522 Neurology Laboratory, Level 6 (South Block), University of Malaya Medical Centre, Kuala Lumpur 50603, Malaysia / Tel: +60-3-7949-2891 / Fax: +60- 3-7949-4613 / E-mail: limshenyang@gmail.com
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- We present a case of beta-propeller protein-associated neurodegeneration, a form of neurodegeneration with brain iron accumulation. The patient harbored a novel mutation in the WDR45 gene. A detailed video and description of her clinical condition are provided. Her movement disorder phenomenology was characterized primarily by limb stereotypies and gait dyspraxia. The patient’s disability was advanced by the time iron-chelating therapy with deferiprone was initiated, and no clinical response in terms of cognitive function, behavior, speech, or movements were observed after one year of treatment.
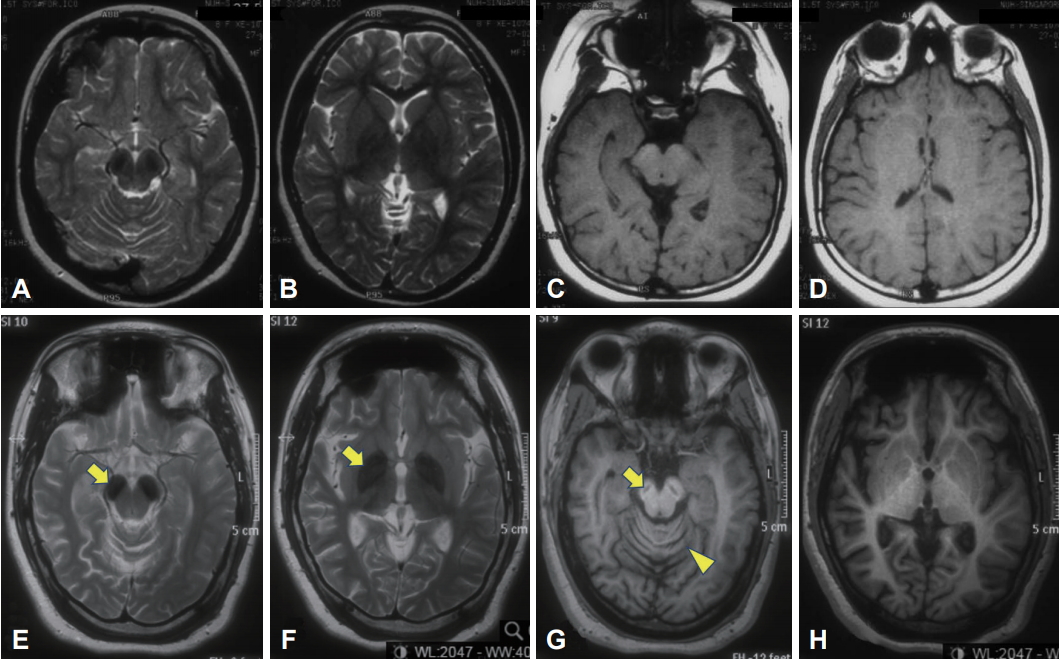
- The patient was the second child of a nonconsanguineous couple. Birth and early developmental history were unremarkable. There was no history of neurological disorders in the immediate or extended family. At 13 months of age, she started having seizures associated with fever and urinary tract infections. The seizures became more frequent during her early childhood and were described as episodes of absence, brief jerks, or sudden forward flexion of the trunk. She became hyperactive with poor attention span and was diagnosed with a learning disability. EEG revealed epileptiform discharges, but brain MRI results were reportedly normal (although in retrospect, bilateral T2 hypointensity could be seen in the substantia nigra) (Figure 1, top panel). She was treated with clonazepam and methylphenidate. At the age of eight, sodium valproate and levetiracetam were added, and the seizures were controlled. She attained her best level of function in her teenage years. She was able to walk independently without a walking aid and even run. She could speak in brief sentences but was never able to read or write.
- In her early 20s, she started to deteriorate with reduced ability to walk and was falling backwards, followed soon after by cognitive decline. Ophthalmological examination revealed optic atrophy without retinal pigmentary changes. EEG showed thetadelta range slowing of the background rhythm with epileptiform discharges. Brain MRI repeated at age 24 (Figure 1, lower panel) showed marked bilateral T2 hypointensity in the substantia nigra and globus pallidus and a “slit” of T1 hypointensity in the nigra with a faint rim of hyperintensity, as well as global cerebral atrophy and cerebellar atrophy. Brain magnetic resonance spectroscopy was normal. Molecular genetic testing revealed a nucleotide change of guanine (G) to adenine (A) at nucleotide position c.249 (c.249G>A) in exon 6 in one copy of the WDR45 gene (accession number NM 007075.3) (Knight Diagnostic Laboratories, Knight Cancer Institute, Oregon Health and Science University, Portland, OR, USA). This variant is not annotated in any published database, including Exome Aggregation Consortium (ExAC), 1000 Genomes, or the National Heart, Lung and Blood Institute Exome Sequencing Project Exome Variant Server (EVS). This mutation predicts an amino acid change of tryptophan (Trp) to a stop codon at codon p.83 (p.Trp83X) (accession number NP 009006) within the WD40 repeat domain of WDR45. Additionally, this mutation is predicted to be pathogenic, potentially resulting in a truncated WDR45 protein, which lacks amino acids 84–361 [5]. Proteomic analysis may be explored in the future to confirm the truncation. Both parents tested negative for the mutation, and screening of an additional 20 control samples from the local population was negative. Her older sister, who lives overseas, also had a negative genetic test result. The patient’s other blood investigations were normal, including blood film (no acanthocytes) and lactate.
- Upon treatment with sodium valproate (200 mg bid), levetiracetam (500 mg bid), baclofen (10 mg bid), trihexyphenidyl (1.5 mg daily), and clonazepam (0.5 mg bid), her seizures were controlled, and episodic limb muscle spasms were reduced. Previous trials of levodopa treatment resulted in generalized dyskinesia and had to be stopped. Her condition when age 28–29 is shown in the video and described in the video legend (please see Supplementary Video 1 in the online-only data supplement).
- A trial of deferiprone, an iron-chelating agent able to cross the blood-brain barrier, was initiated at a dose of 250 mg bid (body weight 65 kg). When this was uptitrated to 500 mg bid, i.e. approximately 15 mg/kg/day (a moderate 20–30 mg/kg/day dose has been suggested for chelation in neurodegenerative disorders, whereas typically approximately 80 mg/kg/day is used to treat iron overload due to blood transfusion in thalassemia) [6-8], the patient experienced distressing abdominal pain, anorexia, insomnia, restlessness, and agitation, and the treatment had to be withheld. Deferiprone was subsequently resumed and gradually increased to 250 mg bid, but she again suffered the same side effects as before. Thus, the dosage had to be reduced back to 250 mg daily. After treatment with deferiprone for a year, there has been no apparent clinical improvement in terms of cognitive function, behavior, speech, or movements. The patient’s symptomatic treatments remained stable over this period. Repeat brain imaging was not performed, as this would have necessitated administration of general anesthesia.
- Interestingly, iron indices prior to initiation of deferiprone showed low systemic iron stores with transferrin saturation of 10% (reference range 20–45%); the serum ferritin level (52.9 μg/L, reference range 10.0–291.0 μg/L), hemoglobin level (121 g/L), and mean corpuscular volume (81 fL) were in the lower range of normal. There was no history of menorrhagia or other overt blood loss, and serum vitamin B12 and folate levels were in the normal range. We were concerned about the possibility of inducing global iron depletion in a patient who was already mildly hypoferremic; however, her transferrin saturation and ferritin levels (ranged between 19–20% and 53.7–67 μg/L, respectively) did not drop further during deferiprone treatment .
CASE REPORT
- We report a case of BPAN with a novel mutation in the WDR45 gene with illustrative video and detailed description of the movement disorder phenomenology, characterized primarily by limb stereotypies and gait dyspraxia. Sequential brain imaging showed evolution of iron deposition and atrophy over time, with involvement of the substantia nigra occurring earlier and more prominently than in the pallidum, as previously described [1].
- Chelation therapy with deferiprone to remove excessive iron in the brain has shown some promise in pantothenate kinase-associated neurodegeneration (PKAN), the most common form of NBIA. In one study (n = 9), treatment for 6 months resulted in improvement of radiological but not clinical status; the authors noted that the majority of their patients were already in an advanced stage (e.g., wheelchairbound) by the time of treatment initiation [7]. Another cohort of patients (n = 6) with milder disability overall compared to the aforementioned study completed 3–4 years of treatment. This cohort showed radiological improvement and stabilization of motor symptoms [8]. However, convincing evidence of clinical benefit is lacking. No serious adverse events were reported [7,8]. Unfortunately, although perhaps not unexpectedly since she was already severely disabled by the time of deferiprone initiation, our patient did not show any clinical response to treatment after one year. Furthermore, only a low dose of deferiprone could be used due to poor patient tolerance. We are aware of only one other published case report of a patient with BPAN (with much milder motoric impairment compared to our patient) who was treated with deferiprone at a relatively high dose of 1,000 mg bid for four months. The treatment coincided with worsening of the patient’s parkinsonian features and was ceased for this reason [9].
- This case adds to the existing literature on a very rare neurodegenerative disease. It is hoped that greater awareness of the NBIAs, including rare forms such as BPAN, will lead to future advances in the scientific understanding and management of these disabling diseases [10].
DISCUSSION
Supplementary Materials
Supplementary Video Legends
- The authors gratefully acknowledge the patient’s family for their consent and participation in this report, including publication of the video. SAS was supported by the Else Kröner-Fresenius Stiftung.
Acknowledgments
- 1. Gregory A, Kurian MA, Haack T, Hayflick SJ, Hogarth P. Beta-propeller protein-associated neurodegeneration. [cited 2017 Mar 19]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK424403/?report=printable.
- 2. Nishioka K, Li G, Yoshino H, Li Y, Matsushima T, Takeuchi C, et al. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging 2015;36:2004. e9-2004.e15. Article
- 3. Paudel R, Li A, Wiethoff S, Bandopadhyay R, Bhatia K, de Silva R, et al. Neuropathology of beta-propeller protein associated neurodegeneration (BPAN): a new tauopathy. Acta Neuropathol Commun 2015;3:39.ArticlePubMedPMCPDF
- 4. Yoganathan S, Arunachal G, Sudhakar SV, Rajaraman V, Thomas M, Danda S. Beta propellar protein-associated neurodegeneration: a rare cause of infantile autistic regression and intracranial calcification. Neuropediatrics 2016;47:123–127.ArticlePubMedPDF
- 5. Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012;91:1144–1449.ArticlePubMedPMC
- 6. Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood 2007;110:401–408.ArticlePubMed
- 7. Zorzi G, Zibordi F, Chiapparini L, Bertini E, Russo L, Piga A, et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: results of a phase II pilot trial. Mov Disord 2011;26:1756–1759.ArticlePubMed
- 8. Cossu G, Abbruzzese G, Matta G, Murgia D, Melis M, Ricchi V, et al. Efficacy and safety of deferiprone for the treatment of pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA): results from a four years follow-up. Parkinsonism Relat Disord 2014;20:651–654.ArticlePubMed
- 9. Fonderico M, Laudisi M, Andreasi NG, Bigoni S, Lamperti C, Panteghini C, et al. Patient affected by beta-propeller protein-associated neurodegeneration: a therapeutic attempt with iron chelation therapy. Front Neurol 2017;8:385.ArticlePubMedPMC
- 10. Jinnah HA, Albanese A, Bhatia KP, Cardoso F, Da Prat G, de Koning TJ, et al. Treatable inherited rare movement disorders. Mov Disord 2018;33:21–35.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Lipid droplet accumulation in Wdr45-deficient cells caused by impairment of chaperone-mediated autophagic degradation of Fasn
Qiuhong Xiong, Huimin Sun, Yanlin Wang, Qian Xu, Yu Zhang, Mei Xu, Zhonghua Zhao, Ping Li, Changxin Wu
Lipids in Health and Disease.2024;[Epub] CrossRef - Quantitative retrospective natural history modeling of WDR45-related developmental and epileptic encephalopathy – a systematic cross-sectional analysis of 160 published cases
Afshin Saffari, Julian Schröter, Sven F. Garbade, Julian E. Alecu, Darius Ebrahimi-Fakhari, Georg F. Hoffmann, Stefan Kölker, Markus Ries, Steffen Syrbe
Autophagy.2022; 18(7): 1715. CrossRef - Cerebral Iron Deposition in Neurodegeneration
Petr Dusek, Tim Hofer, Jan Alexander, Per M. Roos, Jan O. Aaseth
Biomolecules.2022; 12(5): 714. CrossRef - Interactions of dopamine, iron, and alpha-synuclein linked to dopaminergic neuron vulnerability in Parkinson's disease and Neurodegeneration with Brain Iron Accumulation disorders
Rachel M. Wise, Annika Wagener, Urban M. Fietzek, Thomas Klopstock, Eugene V. Mosharov, Fabio A. Zucca, David Sulzer, Luigi Zecca, Lena F. Burbulla
Neurobiology of Disease.2022; 175: 105920. CrossRef -
WDR45 variants cause ferrous iron loss due to impaired ferritinophagy associated with nuclear receptor coactivator 4 and WD repeat domain phosphoinositide interacting protein 4 reduction
Kiwako Tsukida, Shin-ichi Muramatsu, Hitoshi Osaka, Takanori Yamagata, Kazuhiro Muramatsu
Brain Communications.2022;[Epub] CrossRef - Iron Chelation in Movement Disorders: Logical or Ironical
Dinkar Kulshreshtha, Jacky Ganguly, Mandar Jog
Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques.2021; : 1. CrossRef - Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders
Vassilena Iankova, Ivan Karin, Thomas Klopstock, Susanne A. Schneider
Frontiers in Neurology.2021;[Epub] CrossRef - Consensus clinical management guideline for beta‐propeller protein‐associated neurodegeneration
Jenny L Wilson, Allison Gregory, Manju A Kurian, Ittai Bushlin, Fanny Mochel, Lisa Emrick, Laura Adang, Penelope Hogarth, Susan J Hayflick
Developmental Medicine & Child Neurology.2021; 63(12): 1402. CrossRef - WDR45, one gene associated with multiple neurodevelopmental disorders
Yingying Cong, Vincent So, Marina A. J. Tijssen, Dineke S. Verbeek, Fulvio Reggiori, Mario Mauthe
Autophagy.2021; 17(12): 3908. CrossRef - Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation
Robert V.V. Spaull, Audrey K.S. Soo, Penelope Hogarth, Susan J. Hayflick, Manju A. Kurian
Tremor and Other Hyperkinetic Movements.2021;[Epub] CrossRef - The roles of iron and HFE genotype in neurological diseases
Yunsung Kim, James R. Connor
Molecular Aspects of Medicine.2020; 75: 100867. CrossRef - The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System
Karina Joppe, Anna-Elisa Roser, Fabian Maass, Paul Lingor
Frontiers in Neuroscience.2019;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite