E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 9(1); 2016 > Article
-
Original Article
Clinical Heterogeneity of Atypical Pantothenate Kinase-Associated Neurodegeneration in Koreans - Jae-Hyeok Lee1, Jongkyu Park2, Ho-Sung Ryu3, Hyeyoung Park4, Young Eun Kim5, Jin Yong Hong6, Sang Ook Nam7, Young-Hee Sung8, Seung-Hwan Lee9, Jee-Young Lee10, Myung Jun Lee11, Tae-Hyoung Kim12, Chul Hyoung Lyoo13, Sun Ju Chung3, Seong Beom Koh14, Phil Hyu Lee15, Jin Whan Cho2, Mee Young Park16, Yun Joong Kim5, Young H. Sohn15, Beom Seok Jeon4, Myung Sik Lee14
-
Journal of Movement Disorders 2016;9(1):20-27.
DOI: https://doi.org/10.14802/jmd.15058
Published online: January 25, 2016
1Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, Korea
2Department of Neurology, Sungkyunkwan University School of Medicine, Samsung Medical Center, Seoul, Korea
3Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
4Department of Neurology, Seoul National University Hospital, Seoul, Korea
5Department of Neurology, Hallym University Sacred Heart Hospital, Anyang, Korea
6Department of Neurology, Yonsei University Wonju College of Medicine, Wonju, Korea
7Department of Pediatrics, Pusan National University Yangsan Hospital, Yangsan, Korea
8Department of Neurology, Gachon University Gil Hospital, Incheon, Korea
9Department of Neurology, Kangwon National University School of Medicine, Chuncheon, Korea
10Department of Neurology, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul, Korea
11Department of Neurology, Pusan National University Hospital, Busan, Korea
12Department of Neurology, Dong-Eui Hospital, Busan, Korea
13Department of Neurology, Gangnam Severance Hospital, Seoul, Korea
14Department of Neurology, Korea University College of Medicine, Korea University Guro Hospital, Seoul, Korea
15Department of Neurology, Yonsei University College of Medicine, Seoul, Korea
16Department of Neurology, Yeungnam University Medical Center, Daegu, Korea
- Corresponding author: Jae-Hyeok Lee, MD, PhD, Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, 20 Geumo-ro, Mulgeum-eup, Yangsan 50612, Korea / Tel: +82-55-360-2453 / Fax: +82-55-360-2152 / E-mail: jhlee.neuro@pusan.ac.kr
Copyright © 2016 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
-
Objective
- Neurodegeneration with brain iron accumulation (NBIA) represents a group of inherited movement disorders characterized by iron accumulation in the basal ganglia. Recent advances have included the identification of new causative genes and highlighted the wide phenotypic variation between and within the specific NBIA subtypes. This study aimed to investigate the current status of NBIA in Korea.
-
Methods
- We collected genetically confirmed NBIA patients from twelve nationwide referral hospitals and from a review of the literature. We conducted a study to describe the phenotypic and genotypic characteristics of Korean adults with atypical pantothenate kinase-associated neurodegeneration (PKAN).
-
Results
- Four subtypes of NBIA including PKAN (n = 30), PLA2G6-related neurodegeneration (n = 2), beta-propeller protein-associated neurodegeneration (n = 1), and aceruloplasminemia (n = 1) have been identified in the Korean population. The clinical features of fifteen adults with atypical PKAN included early focal limb dystonia, parkinsonism-predominant feature, oromandibular dystonia, and isolated freezing of gait (FOG). Patients with a higher age of onset tended to present with parkinsonism and FOG. The p.R440P and p.D378G mutations are two major mutations that represent approximately 50% of the mutated alleles. Although there were no specific genotype-phenotype correlations, most patients carrying the p.D378G mutation had a late-onset, atypical form of PKAN.
-
Conclusions
- We found considerable phenotypic heterogeneity in Korean adults with atypical PKAN. The age of onset may influence the presentation of extrapyramidal symptoms.
- This study was approved by the Ethical Committee of the Pusan National University Yangsan Hospital. We recruited thirty-four Korean patients with genetically determined NBIA using a structural survey and a literature review of published case reports. A nationwide survey was organized by the Korean Movement Disorders Society. For PKAN, nine new cases and twenty-one previously published cases from twenty-eight families were collected. Among these cases, sixteen patients were classified as having an atypical disease according to the criteria of Hayflick et al. [6]. Three pediatric patients who had typical clinical features of PKAN and the eye-of-the-tiger sign carried only one heterozygous missense mutation that was detected by direct sequencing of the PANK2 gene. Two siblings with PLAN and one patient with beta-propeller protein-associated neurodegeneration (BPAN) were recently described in the literature [16,18]. One unpublished case of aceruloplasminemia, a 60-year-old female presenting with dystonia, chorea, and dementia, was included in the present study.
- We collected detailed clinical data on fifteen adult PKAN patients from fourteen unrelated families. Genomic DNA from all patients was sequenced for PANK2 mutations. For the numbering of the mutations, all published and unpublished cases were adjusted by comparison with the wild-type nucleotide and amino acid sequences (GenBank ID: NM_153638.2, NP_705902.2). For analysis of the clinical and genetic data, the medical records of five new patients from 4 referral hospitals were reviewed. We also attempted to contact the authors of the publications to update the clinical information, and we obtained additional follow-up data from six published cases [8,12-15]. We analyzed the following clinical information: age at onset of the disease, age at diagnosis, age at the last evaluation, presenting symptoms, and clinical characteristics at the last evaluation including dystonia, tremor, parkinsonism, gait/posture, pyramidal signs, bulbar symptoms, cognitive/behavioral changes, results of ophthalmologic examinations, peripheral blood smears, neuroimaging findings, mutations in the PANK2 gene, response to treatment, and disease course. Fourteen pediatric patients with typical PKAN were excluded from this clinical data analysis because of insufficient clinical or genetic information. Instead, we gathered genetic data from ten unrelated pediatric PKAN patients carrying two mutated alleles to investigate the differences in the mutation frequencies between the classic and atypical forms.
MATERIALS & METHODS
- The clinical, neuroimaging and molecular results from fifteen patients (thirteen males and two females with the atypical form of PKAN) are summarized in Table 1 and Supplementary Table 1 (in the online-only Data Supplement). The mean age at onset was 31.1 ± 11.8 years (range 15–52 years), and the mean disease duration was 13.8 ± 9.7 years (range 1–34 years). Patients 7 (P7) and 8 (P8) were siblings. Five patients declared that they had at least 1 family member who was affected with similar symptoms. The remaining patients were presumed to be sporadic cases. The presenting symptoms were as follows: difficulty in writing or handling objects in ten patients (hand dystonia in six patients and hand tremor in four patients), gait disturbance in four patients [dystonic gait in two patients, dystonic gait with freezing in one patient, and isolated freezing of gait (FOG) in one patient], speech changes in three patients (monotonous speech, dysphonia, and dysarthria), and oromandibular dystonia (OMD) in one patient (jaw-opening). Two patients presented with a rapid onset, OMD and painful dystonia (P2 and P7).
- At the last evaluation, the predominant neurological features were dystonia and parkinsonism (eight patients with dystonia without parkinsonism, four patients with parkinsonism and dystonia, and two patients with parkinsonism without dystonia). Dystonia most commonly involved the limbs (n = 12), often affecting the craniocervical musculature including blepharospasm, cervical dystonia, and OMD (n = 8). Early focal limb dystonia progressed to involve other limbs. Axial involvement, with retrocollis or truncal tilting, was not prominent in most of the patients. Gait and balance problems were very common (n = 12), unsteadiness and freezing were reported in six and three patients, respectively. Although some patients needed to use canes for walking, none of them completely lost the ability to walk. Speech disturbances, including dysarthria, palilalia and hypophonia, were frequent (n = 10) and presented from the early stage in three patients. Dysarthria was accompanied by dysphagia or drooling in four patients. Tremors (n = 9) were usually postural in nature and presented as dystonic tremors. Rest tremors were observed in two patients; one of them presented with parkinsonism (P13), and the other had unilateral leg tremors (P12). Corticospinal tract signs (spasticity, hyperreflexia and extensor plantar reflexes) were rarely described.
- Other features including pigmentary retinopathy, acanthocytosis [19], and seizure were not mentioned in the medical records. Psychiatric symptoms with cognitive decline were not prominent in most patients. Cognitive impairment was evident in two young siblings (P7 and P8). The results of neuropsychological tests revealed mild mental retardation. The older brother showed psychiatric signs of obsessive-compulsive disorder and attention-deficit and hyperactivity disorder. Two patients who underwent neuropsychological testing had mild frontal dysfunction (P10 and P15). Three patients over 50 years of age presented with subjective memory complaints; however, neuropsychological tests were not performed.
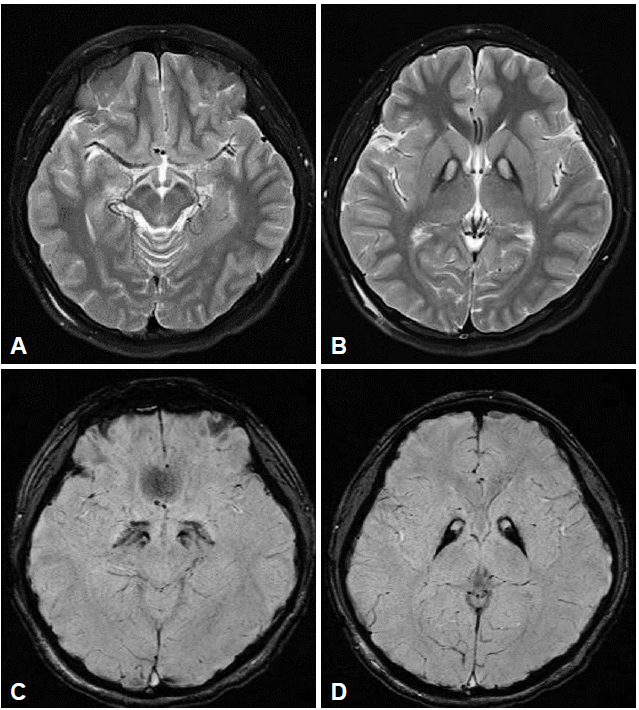
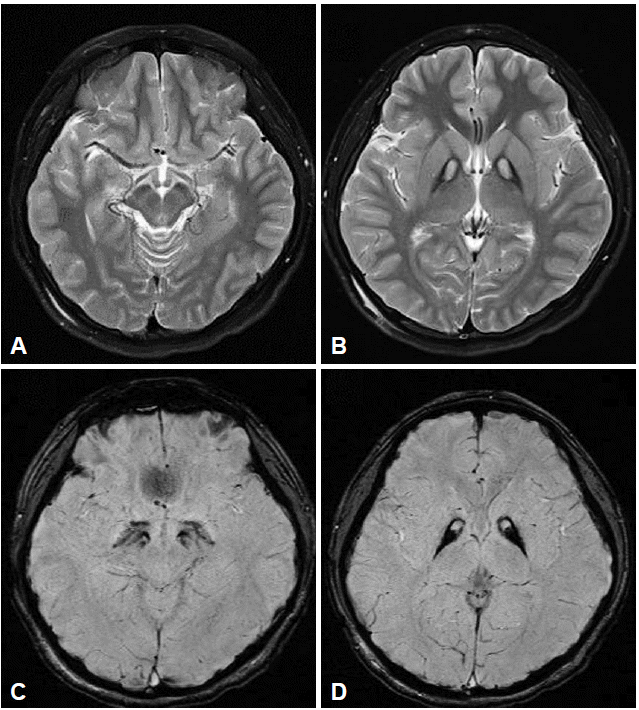
- All had the eye-of-the-tiger sign on T2-weighted images. Marked hyperintensities on T1-weighted images in bilateral anteromedial globus pallidi were reported in two patients (P5 and P10). Susceptibility-weighted imaging dramatically revealed the evidence of iron deposition restricted to the globus pallidus and substantia nigra (Figure 1). Dopamine transporter imaging with N-{3-[(18)F]fluoropropyl}-2-carbomethoxy-3-(4-iodophenyl) nortropane positron emission tomography studies in two patients with parkinsonism (P13) and gait freezing (P15) revealed no striatal dopamine depletion. No perfusion defect was observed in one patient (P3).
- There were no overt responses to conventional drugs used for the control of dystonia, tremor, or parkinsonism, although partial improvements were reported. In two patients with FOG as the main clinical feature, FOG showed dramatic responses to medications [7,17]. One patient (P9) dramatically improved with trihexyphenidyl, and the other patient (P15) dramatically improved with methylphenidate. Three patients (P4, P7, and P10) underwent bilateral globus pallidus pars interna (GPi) deep brain stimulation (DBS). All patients showed some improvements in the clinical rating scales of dystonia and disability (Supplementary Table 1 in the online-only Data Supplement). The patient with rapid exacerbation of painful dystonic spasms immediately underwent bilateral GPi-DBS (P7). His axial and limb dystonia showed sustained improvement for up to 5 years after DBS. However, he developed intractable jaw-opening OMD which severely interfered with speech and mastication. In two other patients (P4 and P10), disabling hand dystonia responded to DBS. Their functional improvement continued for 3-year follow-up period. However, in one patient (P4, previously published), the effect disappeared four year after DBS.
- We identified 18 different mutations in 24 Korean families with PKAN. Mutations were found in both PANK2 alleles and included missense mutations (37/48 alleles), frameshift mutations (8/48 alleles), nonsense mutations (1/48 alleles), and deletions (4/48 alleles) (Table 2). Most of the patients had compound heterozygous mutations. In two atypical cases, one was homozygous (P12), and the other had two different missense mutations and one frameshift mutation (P13). An intragenic deletion ranging from exons 2 to 4 (c.629-?_1412+ ?del) was found in a pediatric patient [15]. The most frequent mutations identified were p.D378G (10/28 alleles) and p.R440P (8/28 alleles) in Korean adults with atypical PKAN (Table 2: atypical). Within the atypical group, there were no specific genotype-phenotype correlations. Five patients who were all compound heterozygotes for these two mutations (p. D378G and p.R440P) showed variable clinical manifestations, including dystonia without parkinsonism (n = 2), dystonia with parkinsonism (n = 1), parkinsonism-predominant (n = 1), and isolated FOG (n = 1). Two siblings (P7 and P8) presented with different spreading patterns of dystonia, although they had a similar age at onset in the second decade. The older brother, initially, developed twisting postures in his right hand, whereas his younger brother had dystonia starting in the legs.
- In pediatric patients with typical PKAN, the most prevalent mutation was the p.R440P mutation (9/20 alleles), followed by the p.I501N mutation (3/20 alleles) (Table 2: classic). Eight published cases initially presented with gait disturbance and generalized dystonia before 10 years of age. Half of them had lost ambulation within 3 years of onset [15]. Three newly enrolled patients had dystonic opisthotonus and tip-toe gait. Two possibly pathogenic novel variants were identified (c.1676C > G, p.A559G and c.1257del, p.F419fs). One adolescence patient with atypical presentation had two compound heterozygous mutations (c.1351C > T, p.R451X and c.1607A > G, p.Y536C) [15]. The allele frequencies of protein-truncating mutations were not different between the two groups (Table 2).
RESULTS
- Among the ten forms of NBIA, four subtypes of NBIA including PKAN, PLAN, BPAN, and aceruloplasminemia have been identified in the Korean population. PKAN was the most common cause of NBIA. The characteristic eye-of-the-tiger sign of PKAN may aid in diagnosing this disorder [1,6]. The other forms of NBIA were rare and were reported only in single cases [16,18]. However, it may still be too early to determine the frequency and distribution of these disorders in the Korean population. Increasing clinician awareness will improve early recognition and diagnosis of various types of NBIA.
- All fifteen patients analyzed in this study could be classified as having atypical PKAN on the basis of late onset and slow progression [1,6]. Patients with long-lasting movement disorders exhibited similar functional disabilities regardless of the disease duration and no significant functional declines during the past several years. None of them completely lose their ability to ambulate even after a disease duration of 20 years or more. This observation is most likely related to the specific pattern of disease progression of atypical PKAN. The rate of decline tends to be steeper after the onset of symptoms, followed by a relatively stable period of slower progression [1,20].
- In addition to the age at onset and disease duration, other atypical features in our patients included early focal limb dystonia, tremor, parkinsonism, changes in speech patterns, OMD, and FOG. Early dystonia frequently involved a unilateral hand with manifestation of writer’s cramp or action-induced dystonia. Most of them slowly progressed to involve other limbs and, less commonly, craniocervical regions. This initial distribution of dystonia is different from that of patients with the classic form that predominantly affects the lower limbs. Dystonic opisthotonus, a hallmark of early-onset typical PKAN [21], was rare in our series. The presentation and progression of dystonia tends to differ between ethnic groups. As demonstrated by a previous report, OMD was less common than in Caucasians [20,22]. In a recent prospective study, half of the patients developed dystonic opisthotonus within five years after onset [20]. This disease course was more progressive than that observed in our patients.
- The parkinsonism-predominant phenotype was also the characteristic subgroup of our patients. They initially presented with hand tremor, even with a resting component, and were clinically indistinguishable from patients with degenerative parkinsonism. The results of normal striatal binding with dopamine transporter imaging are consistent with previous reports [23]. Moreover, a neuropathological study identified only minimal loss of neuromelanin in the substantia nigra, while the central pallidum and substantia nigra pars reticulata were mainly involved [24]. Two major extrapyramidal symptoms of PKAN, dystonia and parkinsonism, showed age-dependency, as previously reported [1,25]. Patients with parkinsonism tended to have a later age of onset than patients in the dystonia-predominant group, while adolescent patients or patients in early adulthood demonstrated more dystonia than parkinsonism. In addition, though the number of cases was limited, dystonic patients with late-adult onset tended to have less craniocervical involvement at initial presentation. The age of onset may influence the presentation of extrapyramidal symptoms in the atypical form of PKAN.
- Contrary to previous reports [5,6,26], neuropsychiatric symptoms and cognitive impairment were uncommon and were not prominent in our series. This discrepancy can be explained by ethnicity-related differences. A previous comparative analysis noted a lower prevalence of these symptoms in Asians than in Caucasians [22]. Epidemiological surveys showed lower prevalence rates of mental disorders in Asians [27]. However, other confounding factors should be considered. Standardized assessment measures revealed that age of onset was associated with intellectual and adaptive functioning [28]. A younger age at onset of the disease was associated with a greater degree of cognitive impairment [1]. Neuropsychiatric symptoms can be early presenting signs of adolescence [1]. Patients enrolled in this study were much older than those of previous reports [5,6,22,26]. Most of them revealed stable preserved cognition and behavior except for two siblings with an age of onset in the mid-teenage years. Patients who survive into middle or old age should not be assumed to have a progressive dementia [29]. However, they may be underdiagnosed due to insufficient evaluation. Further systematic assessment is warranted to determine the ethnic differences in cognitive and psychiatric symptoms.
- The p.R440P and p.D378G mutations are two major mutations representing approximately 50% of the mutated alleles. Most of the mutations were different from those reported in Caucasians [6,22,26]. The common mutations (p.G521R and p.T528M) found in Caucasians have not been found in any Korean PKAN patients so far. The p.D378G mutation is also a frequent mutation in Chinese individuals and most likely represents a hotspot in PKAN patients from Eastern Asia [22,30]. The p.R440P mutation may be a founder genotype in the Korean population [15]. This mutation was associated with both classic and atypical phenotypes [15]. Notably, most patients carrying the p.D378G mutation had a late-onset, atypical form of PKAN [30]. Previous studies have shown that the locations of the missense mutations correlated with the residual activity of pantothenate kinase and the age of onset [26,31]. The enzymatic activity of the p.D378G mutation should be determined to confirm the association of this mutation with the atypical clinical phenotype. It has been shown that two null mutations result in a loss of enzyme activity and lead to early-onset classic disease [6,26]. However, no Korean pediatric patients with classic PKAN had two predicted loss-of-function alleles. Other genetic modifiers and environmental factors may also be involved.
- Dystonia and parkinsonism showed limited responses to medications. The preservation of nigrostriatal dopaminergic integrity can be associated with limited benefits of dopaminergic drugs. Isolated FOG with drug responsiveness is the most striking presenting feature in our series, although the exact mechanism remains uncertain [7,17]. GPi-DBS performed in three patients was shown to be partially effective for relieving dystonic symptoms. The patients exhibited a sustained positive response to GPi-DBS for a 3- to 5-year follow-up period. Previous studies have demonstrated the efficacy of bilateral GPi-DBD for the treatment of dystonia in patients with PKAN, although the effect was not as great as the benefit reported in patients with primary generalized dystonia [15,32]. However, most reports are limited by short periods of observation. Additional follow-up data in this study add to the evidence supporting the long-term benefit [33].
- In this study, we attempted a more detailed analysis of clinical manifestations in Korean adults with atypical PKAN, and we found considerable phenotypic heterogeneity. Some characteristic symptoms of the atypical form tended to show an age-dependent presentation. However, the present study is limited because every detail of the clinical severities could not be obtained as our data were gathered retrospectively from medical records and from a review of literatures. Clinical rating scales primarily focused on PKAN or well-structured registries should be used for the clearer delineation of the severity and natural history of this disease [1].
DISCUSSION
Supplementary Materials
- This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2014R1A1A2059252) and by the Pusan National University Yangsan Hospital (2015).
Acknowledgments
|
Atypical (n = 15*) |
Classic (n = 10) |
||||||
|---|---|---|---|---|---|---|---|
| Exon | Genomic DNA | Protein | No. | Exon | Genomic DNA | Protein | No. |
| 3 | c.1133A > G | p.D378G | 10 | 4 | c.1319G > C | p.R440P | 9 |
| 4 | c.1319G > C | p.R440P | 8 | 5 | c.1502T > A | p.I501N | 3 |
| 3 | c.1003A > G | p.K335E | 3 | 3 | c.1153delCinsTT | p.L385fs | 1 |
| 4 | c.1273_1275del | p.L425del | 2 | 4 | c.1273_1275del | p.L425del | 1 |
| 3 | c.1153delCinsTT | p.L385fs | 2 | 4 | c.1257del† | p.F419fs | 1 |
| 3 | c.1154_1155insTT | p.L385fs | 1 | 1 | c.391_418del | p.A131fs | 1 |
| 4 | c.1270_1272del | p.L424del | 1 | 7 | c.1676C > G† | p.A559G | 1 |
| 2 | c.823_824del | p.L275fs | 1 | 4 | c.1301C > T | p.A434V | 1 |
| 5 | c.1500_1501del | p.I501fs | 1 | 2 | c.852T > G | p.F284L | 1 |
| 4 | c.1351C > T | p.R451X | 1 | 2 to 4 | c.629-?_1412+ ?del‡ | 1 | |
| 6 | c.1607A > G | p.Y536C | 1 | ||||
- 1. Hogarth P. Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord 2015;8:1–13.ArticlePubMedPMC
- 2. Arber CE, Li A, Houlden H, Wray S. Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol; 2015 Apr. 14. [Epub]. http://dx.doi.org/10.1111/nan.12242. ArticlePubMedPMC
- 3. Schneider SA. Neurodegenerations with brain iron accumulation. Parkinsonism Relat Disord 2016;22 Suppl 1:S21–S25.Article
- 4. Keogh MJ, Chinnery PF. Current concepts and controversies in neurodegeneration with brain iron accumulation. Semin Pediatr Neurol 2012;19:51–56.ArticlePubMed
- 5. Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, Scarano V, et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 2005;64:1810–1812.ArticlePubMed
- 6. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003;348:33–40.ArticlePubMed
- 7. Lyoo CH, Prokisch H, Meitinger T, Lee SY, Kim do H, Lee MS. Anticholinergic-responsive gait freezing in a patient with pantothenate kinase-associated neurodegeneration. Mov Disord 2008;23:283–284.ArticlePubMed
- 8. Chung SJ, Lee JH, Lee MC, Yoo HW, Kim GH. Focal hand dystonia in a patient with PANK2 mutation. Mov Disord 2008;23:466–468.ArticlePubMed
- 9. Lee SY, Lyoo CH, Seo KD, Lee MS. Three patients with classic and atypical neurodegeneration with brain iron accumulation. J Korean Neurol Assoc 2008;26:243–246.
- 10. Seo JH, Song SK, Lee PH. A novel PANK2 mutation in a patient with atypical pantothenate-kinase-associated neurodegeneration presenting with adult-onset parkinsonism. J Clin Neurol 2009;5:192–194.ArticlePubMedPMC
- 11. Kim SH, Sung YH, Park KH, Lee YB, Park HM, Shin DJ, et al. Novel compound heterozygous mutations in the pantothenate kinase 2 gene in a Korean patient with atypical pantothenate kinase associated neurodegeneration. J Mov Disord 2009;2:45–47.ArticlePubMedPMCPDF
- 12. Yoon WT, Lee WY, Shin HY, Lee ST, Ki CS. Novel PANK2 gene mutations in Korean patient with pantothenate kinase-associated neurodegeneration presenting unilateral dystonic tremor. Mov Disord 2010;25:245–247.ArticlePubMed
- 13. Lee JH, Kim DS, Baik SK, Nam SO. Nigropallidal iron accumulation in pantothenate kinase-associated neurodegeneration demonstrated by susceptibility-weighted imaging. J Neurol 2010;257:661–662.ArticlePubMed
- 14. Kim J, Shin H, Youn J, Ki CS, Cho AR, Cho JW. A novel PANK2 gene mutation with sudden-onset dystonia. Can J Neurol Sci 2012;39:395–397.ArticlePubMed
- 15. Lim BC, Ki CS, Cho A, Hwang H, Kim KJ, Hwang YS, et al. Pantothenate kinase-associated neurodegeneration in Korea: recurrent R440P mutation in PANK2 and outcome of deep brain stimulation. Eur J Neurol 2012;19:556–561.ArticlePubMed
- 16. Kim YJ, Lyoo CH, Hong S, Kim NY, Lee MS. Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat Disord 2015;21:402–406.ArticlePubMed
- 17. Kwon KY, Lee HM, Kim M, Kang SH, Koh SB. Long-lasting isolated freezing of gait with good response to methylphenidate: a patient with pantothenate kinase-associated neurodegeneration. Parkinsonism Relat Disord 2015;21:671–672.ArticlePubMed
- 18. Ryu SW, Kim JS, Lee SH. Beta-Propeller-Protein-Associated Neurodegeneration: a case of mutation in WDR45. J Clin Neurol 2015;11:289–291.ArticlePubMedPMC
- 19. Walker RH. Untangling the thorns: advances in the neuroacanthocytosis syndromes. J Mov Disord 2015;8:41–54.ArticlePubMedPMC
- 20. Tomić A, Petrović I, Svetel M, Dobričić V, Dragašević Mišković N, Kostić VS. Pattern of disease progression in atypical form of pantothenate-kinase-associated neurodegeneration (PKAN) - prospective study. Parkinsonism Relat Disord 2015;21:521–524.ArticlePubMed
- 21. Stamelou M, Lai SC, Aggarwal A, Schneider SA, Houlden H, Yeh TH, et al. Dystonic opisthotonus: a “red flag” for neurodegeneration with brain iron accumulation syndromes? Mov Disord 2013;28:1325–1329.ArticlePubMedPMC
- 22. Lee CH, Lu CS, Chuang WL, Yeh TH, Jung SM, Huang CL, et al. Phenotypes and genotypes of patients with pantothenate kinase-associated neurodegeneration in Asian and Caucasian populations: 2 cases and literature review. ScientificWorldJournal 2013;2013:860539.ArticlePubMedPMCPDF
- 23. Cossu G, Cella C, Melis M, Antonini A, Floris GL, Ruffini L, et al. [123I]FP-CIT SPECT findings in two patients with Hallervorden-Spatz disease with homozygous mutation in PANK2 gene. Neurology 2005;64:167–168.ArticlePubMed
- 24. Kruer MC, Hiken M, Gregory A, Malandrini A, Clark D, Hogarth P, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011;134(Pt 4):947–958.ArticlePubMedPMCPDF
- 25. Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord 2004;19:36–42.ArticlePubMed
- 26. Hartig MB, Hörtnagel K, Garavaglia B, Zorzi G, Kmiec T, Klopstock T, et al. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol 2006;59:248–256.ArticlePubMed
- 27. Sue S, Yan Cheng JK, Saad CS, Chu JP. Asian American mental health: a call to action. Am Psychol 2012;67:532–544.ArticlePubMedPDF
- 28. Freeman K, Gregory A, Turner A, Blasco P, Hogarth P, Hayflick S. Intellectual and adaptive behaviour functioning in pantothenate kinase-associated neurodegeneration. J Intellect Disabil Res 2007;51(Pt 6):417–426.ArticlePubMedPMC
- 29. Nicholas AP, Earnst KS, Marson DC. Atypical Hallervorden-Spatz disease with preserved cognition and obtrusive obsessions and compulsions. Mov Disord 2005;20:880–886.ArticlePubMed
- 30. Ma LY, Wang L, Yang YM, Lu Y, Cheng FB, Wan XH. Novel gene mutations and clinical features in patients with pantothenate kinase-associated neurodegeneration. Clin Genet 2015;87:93–95.ArticlePubMed
- 31. Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VM. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci 2005;25:689–698.ArticlePubMedPMC
- 32. Timmermann L, Pauls KA, Wieland K, Jech R, Kurlemann G, Sharma N, et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain 2010;133(Pt 3):701–712.ArticlePubMedPMCPDF
- 33. Krause M, Fogel W, Tronnier V, Pohle S, Hörtnagel K, Thyen U, et al. Long-term benefit to pallidal deep brain stimulation in a case of dystonia secondary to pantothenate kinase-associated neurodegeneration. Mov Disord 2006;21:2255–2257.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Typical pantothenate kinase-associated neurodegeneration caused by compound heterozygous mutations in PANK2 gene in a Chinese patient: a case report and literature review
Yilun Tao, Chen Zhao, Dong Han, Yiju Wei, Lihong Wang, Wenxia Song, Xiaoze Li
Frontiers in Neurology.2023;[Epub] CrossRef - The first Vietnamese patient who presented late onset of pantothenate kinase-associated neurodegeneration diagnosed by whole exome sequencing: A case report
Van Khanh Tran, Chi Dung Vu, Hai Anh Tran, Nguyen Thi Kim Lien, Nguyen Van Tung, Nguyen Ngoc Lan, Huy Thinh Tran, Nguyen Huy Hoang
Medicine.2023; 102(43): e34853. CrossRef - Genetic mutation spectrum of pantothenate kinase-associated neurodegeneration expanded by breakpoint sequencing in pantothenate kinase 2 gene
Dahae Yang, Sanghyun Cho, Sung Im Cho, Manjin Kim, Moon-Woo Seong, Sung Sup Park
Orphanet Journal of Rare Diseases.2022;[Epub] CrossRef - Long-Term Outcomes of Deep Brain Stimulation in Pantothenate Kinase-Associated Neurodegeneration-Related Dystonia
Kyung Ah Woo, Han-Joon Kim, Seung-Ho Jeon, Hye Ran Park, Kye Won Park, Seung Hyun Lee, Sun Ju Chung, Jong-Hee Chae, Sun Ha Paek, Beomseok Jeon
Journal of Movement Disorders.2022; 15(3): 241. CrossRef - Psychiatric symptoms in an adolescent reveal a novel compound heterozygous mutation of the PANK2 gene in the atypical PKAN syndrome
Luz María González Huerta, Sorina Gómez González, Jaime Toral López
Psychiatric Genetics.2021; 31(3): 95. CrossRef - Rational Design of Novel Therapies for Pantothenate Kinase–Associated Neurodegeneration
Nivedita Thakur, Thomas Klopstock, Suzanne Jackowski, Enej Kuscer, Fernando Tricta, Aleksandar Videnovic, Hyder A. Jinnah
Movement Disorders.2021; 36(9): 2005. CrossRef - Atypical Pantothenate Kinase-Associated Neurodegeneration with variable phenotypes in an Egyptian family
Ali S. Shalash, Thomas W. Rösler, Ibrahim Y. Abdelrahman, Hatem S. Abulmakarem, Stefanie H. Müller, Franziska Hopfner, Gregor Kuhlenbäumer, Günter U. Höglinger, Mohamed Salama
Heliyon.2021; : e07469. CrossRef - Treatment Responsiveness of Parkinsonism in Atypical Pantothenate Kinase‐Associated Neurodegeneration
Jeanne Feuerstein, Caroline Olvera, Michelle Fullard
Movement Disorders Clinical Practice.2020;[Epub] CrossRef - Diagnostic and clinical experience of patients with pantothenate kinase-associated neurodegeneration
Randall D. Marshall, Abigail Collins, Maria L. Escolar, H. A. Jinnah, Thomas Klopstock, Michael C. Kruer, Aleksandar Videnovic, Amy Robichaux-Viehoever, Colleen Burns, Laura L. Swett, Dennis A. Revicki, Randall H. Bender, William R. Lenderking
Orphanet Journal of Rare Diseases.2019;[Epub] CrossRef - Intrafamilial variability and clinical heterogeneity in a family with PLA2G6-associated neurodegeneration
Jong Kyu Park, Jinyoung Youn, Jin Whan Cho
Precision and Future Medicine.2019; 3(3): 135. CrossRef - On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation
C. Tello, A. Darling, V. Lupo, B. Pérez‐Dueñas, C. Espinós
Clinical Genetics.2018; 93(4): 731. CrossRef - Looking Deep into the Eye-of-the-Tiger in Pantothenate Kinase–Associated Neurodegeneration
J.-H. Lee, A. Gregory, P. Hogarth, C. Rogers, S.J. Hayflick
American Journal of Neuroradiology.2018; 39(3): 583. CrossRef - Parkinson’s Disease and Metal Storage Disorders: A Systematic Review
Edward Botsford, Jayan George, Ellen Buckley
Brain Sciences.2018; 8(11): 194. CrossRef - Atypical pantothenate kinase-associated neurodegeneration: Clinical description of two brothers and a review of the literature
S. Mahoui, A. Benhaddadi, W. Ameur El Khedoud, M. Abada Bendib, M. Chaouch
Revue Neurologique.2017; 173(10): 658. CrossRef - Clinical rating scale for pantothenate kinase‐associated neurodegeneration: A pilot study
Alejandra Darling, Cristina Tello, María Josep Martí, Cristina Garrido, Sergio Aguilera‐Albesa, Miguel Tomás Vila, Itziar Gastón, Marcos Madruga, Luis González Gutiérrez, Julio Ramos Lizana, Montserrat Pujol, Tania Gavilán Iglesias, Kylee Tustin, Jean Pie
Movement Disorders.2017; 32(11): 1620. CrossRef - Missions of <italic>Journal of Movement Disorders</italic>
Yun Joong Kim
Journal of Movement Disorders.2016; 9(1): 1. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite