Mechanism of Anti-α-Synuclein Immunotherapy
Article information
Abstract
Immunization therapy targeting α-synuclein has emerged as a promising approach for Parkinson’s disease and perhaps for other synucleinopathies. Several antibodies have shown therapeutic effects in mouse models of synucleinopathies and have alleviated the pathological and behavioral phenotypes of these mice. The mechanisms through which the immunization therapy works were initially puzzling, especially given that α-synuclein is a typical cytosolic protein. Recent studies, however, suggested that extracellular α-synuclein is an important pathogenic entity, and hence, a target for immunotherapy. Here, we review the literature describing immunization therapy for synucleinopathies in mouse models and provide current thoughts on the potential mechanisms underlying the therapeutic effects of α-synuclein immunotherapy.
INTRODUCTION
α-synuclein is a neuronal protein highly expressed in presynaptic terminals and is thought to be involved in the regulation of synaptic functions [1]. More attention has been placed on the pathogenic functions of this protein in a group of neurodegenerative diseases referred to as synucleinopathies. These diseases, which include Parkinson’s disease (PD), dementia with Lewy bodies, multiple system atrophy, and a large proportion of Alzheimer’s disease, are pathologically characterized by abnormal accumulation of α-synuclein aggregates. The link between α-synuclein and disease is particularly strong in PD, in which aggregated α-synuclein accumulates in structures known as Lewy bodies and Lewy neurites [2]. Several missense mutations [3-7] and gene multiplication mutations [8-11] in SNCA, the gene for α-synuclein, have been linked to familial forms of PD. Furthermore, SNCA is the gene that is most strongly and consistently associated with sporadic PD [12,13].
PD is clinically characterized by parkinsonian motor symptoms, including resting tremor, muscle tone rigidity, bradykinesia, and postural instability [14]. However, PD patients also manifest a variety of non-motor symptoms, such as autonomic dysfunctions, sensory abnormalities, psychiatric symptoms, sleep disorders, and dementia [15]. The majority of PD patients develop these symptoms sequentially as the disease progresses. Strikingly, an analysis of Lewy bodies revealed a progressive spreading of α-synuclein aggregates with disease progression, and the pattern in which the aggregates spread through the brain seemed to correlate with the clinical progression of the disease [16]. These findings strongly suggest that the spread of α-synuclein aggregates drives the disease progression, and therefore, stopping the spread of α-synuclein aggregates might halt the disease progression. Recent studies provide strong evidence that cell-to-cell propagation of α-synuclein aggregates is the underlying mechanism for the spreading of Lewy pathology [17].
Studies during the past twenty years testify to the importance of α-synuclein and its aggregation in the initiation and progression of PD, and probably other synucleinopathies, making this protein the most promising therapeutic target for these diseases. However, α-synuclein-targeting drugs have yet to be developed. In this review, we propose that immunotherapy for α-synuclein might be a promising approach for developing anti-synucleinopathy therapy and explain how this approach might work mechanistically.
ACTIVE AND PASSIVE IMMUNIZATION OF THE SYNUCLEINOPATHY MODEL MICE
In recent years, immunotherapy has emerged as a promising approach for targeting and clearing protein aggregate pathology in neurodegenerative diseases [18-22]. In a study performed ten years ago, which assessed the feasibility of PD immunotherapy, a transgenic mouse model for synucleinopathies was actively immunized with recombinant α-synuclein protein. The mice successfully generated antibodies against α-synuclein, and the behavioral deficits, α-synuclein deposition and neurodegeneration in the brains of these mice were significantly ameliorated [23]. Likewise, passive immunization with a monoclonal antibody with the epitope of the C-terminal part of α-synuclein decreased the accumulation of α-synuclein aggregates, as well as reduced the behavioral deficits in an α-synuclein transgenic mouse model [24]. Interestingly, administration of antibodies against α-synuclein oligomers reduced α-synuclein levels in both cell lysates and conditioned media [25].
Initially, the effects of immunization in the synucleinopathy models were puzzling and unexplainable, given the cytosolic nature of the target protein [26]; no rational explanation could be provided for how antibodies access α-synuclein proteins. In the following sections, we will discuss recent progress toward resolving this issue.
EXTRACELLULAR α-SYNUCLEIN
Secretion of α-synuclein from neuronal cells
α-synuclein is a typical cytosolic protein and is mostly present in the cytosolic fractions of brain homogenates and neuronal cell homogenates. However, a small portion of cellular α-synuclein is present in the lumen of vesicles [27], the identity of which is yet to be elucidated. These vesicular α-synuclein proteins were secreted from neuronal cells through unconventional exocytosis [28], which collectively refers to endoplasmic reticulum/Golgi-independent exocytosis. The precise mechanism of the exocytosis, however, is unknown. Recently, exosome-associated exocytosis [29] and exophagy (autophagosome-mediated exocytosis) [30] have been suggested as the mechanisms underlying α-synuclein secretion. However, the results of some studies contradict these proposals [31], and the amount of secreted α-synuclein that is associated with extracellular vesicles explains only a very small fraction of the total amount of α-synuclein secreted.
Although the mechanisms of exocytosis are unknown, we do know several conditions under which α-synuclein secretion is enhanced. These conditions, which include proteasome inhibition [28], lysosomal inhibition [32], autophagy inhibition [33], mitochondrial inhibition, oxidative modifications [34,35], and heat shock [29] which commonly affect cellular proteostasis (protein folding homeostasis). A large portion of secreted α-synuclein is oligomeric, whereas the cytosolic α-synuclein is mostly monomers [36]. From these results, we speculate that exocytosis of α-synuclein, and perhaps many other proteins that go through the same pathway, is part of the cellular response to the misfolding of the protein. More work needs to be done to resolve this problem.
Pathogenic actions of extracellular α-synuclein
After secretion from neuronal cells, α-synuclein can act on neighboring cells. Extracellular α-synuclein can be internalized into neuronal cells [37-39]. These proteins undergo endosomal trafficking [37-39] and are delivered to lysosomes where they are degraded [40]. If the internalized α-synuclein can survive the lysosomal degradation, which could result from lysosomal dysfunction, it can induce aggregation of endogenous α-synuclein proteins. Under certain conditions, this aggregate transmission coincides with neuronal cell death, both in cell cultures and in vivo [38,41,42]. However, neurodegeneration does not always occur with aggregate transmission [39,43].
Extracellular α-synuclein also acts on glial cells. α-synuclein released from neurons is transferred to astrocytes, where it induces pro-inflammatory responses [44]. Extracellular α-synuclein can also activate microglia and subsequently trigger inflammatory responses. In microglia, oligomeric forms of neuronreleased α-synuclein interact with toll-like receptor 2 (TLR2) and activate the TLR2 signaling pathway [36]. The interaction between TLR2 and α-synuclein seems to be highly conformation-selective; only certain types of oligomers interact with and activate TLR2, whereas monomers, fibrils, and some oligomer types do not. There seem to be other receptors for α-synuclein in microglia. One of these receptors might be β1-integrin, the activation of which is responsible for the morphological changes and migration of microglia [45].
Although extracellular α-synuclein accounts for only a minor portion of the total brain α-synuclein, its actions on neighboring neurons and glia suggest that it might be an excellent therapeutic target for PD and other synucleinopathies.
MECHANISMS UNDERLYING ANTI-α-SYNUCLEIN IMMUNOTHERAPY
The mechanisms underlying the therapeutic effects of immunization against α-synuclein remain elusive. It has become increasingly clear that extracellular α-synuclein itself and the cellular events that lead to its generation and clearance are promising therapeutic targets for PD. Here, we discuss two mechanisms through which immunotherapy might work.
Clearance of extracellular α-synuclein
Extracellular α-synuclein aggregates can be internalized to neurons and glia. Among the cell types that can internalize α-synuclein aggregates, microglia exhibit the most rapid clearance of these proteins [46]. More recently, it was shown that antibody assisted in the clearance of extracellular α-synuclein, resulting in reduced neuronal and glial accumulation of α-synuclein [47]. This antibody treatment also ameliorated neurodegeneration and behavioral deficits in a mouse model of synucleinopathy. That study showed that microglia became better scavengers for extracellular α-synuclein aggregates in the presence of specific antibodies against α-synuclein. Antibody-α-synuclein immune complexes entered microglia through the Fcγ receptors, which led to efficient delivery of these immune complexes to lysosomes, hence resulting in fast degradation. By clearing extracellular α-synuclein, antibody treatment significantly reduced the extent of cell-to-cell transmission of the protein in mouse models, suggesting that this approach is effective in slowing the disease progression. Furthermore, clinical advantages might be expected if extracellular α-synuclein is selectively targeted by immunotherapy with the intraneuronal α-synuclein being left intact.
Physical blocking of extracellular α-synuclein
In addition to the accelerated clearance of extracellular α-synuclein, antibodies could directly block cell-to-cell transmission of the protein. Tran et al. [48] recently showed that several antibodies against α-synuclein physically blocked intercellular transmission of α-synuclein aggregates in cell cultures and animal models. Therefore, from the results thus far, we propose a working model in which antibodies capture extracellular α-synuclein aggregates and physically interfere with the transfer of the protein to neighboring cells, thus enhancing the efficiency of its uptake into microglia for clearance (Figure 1).
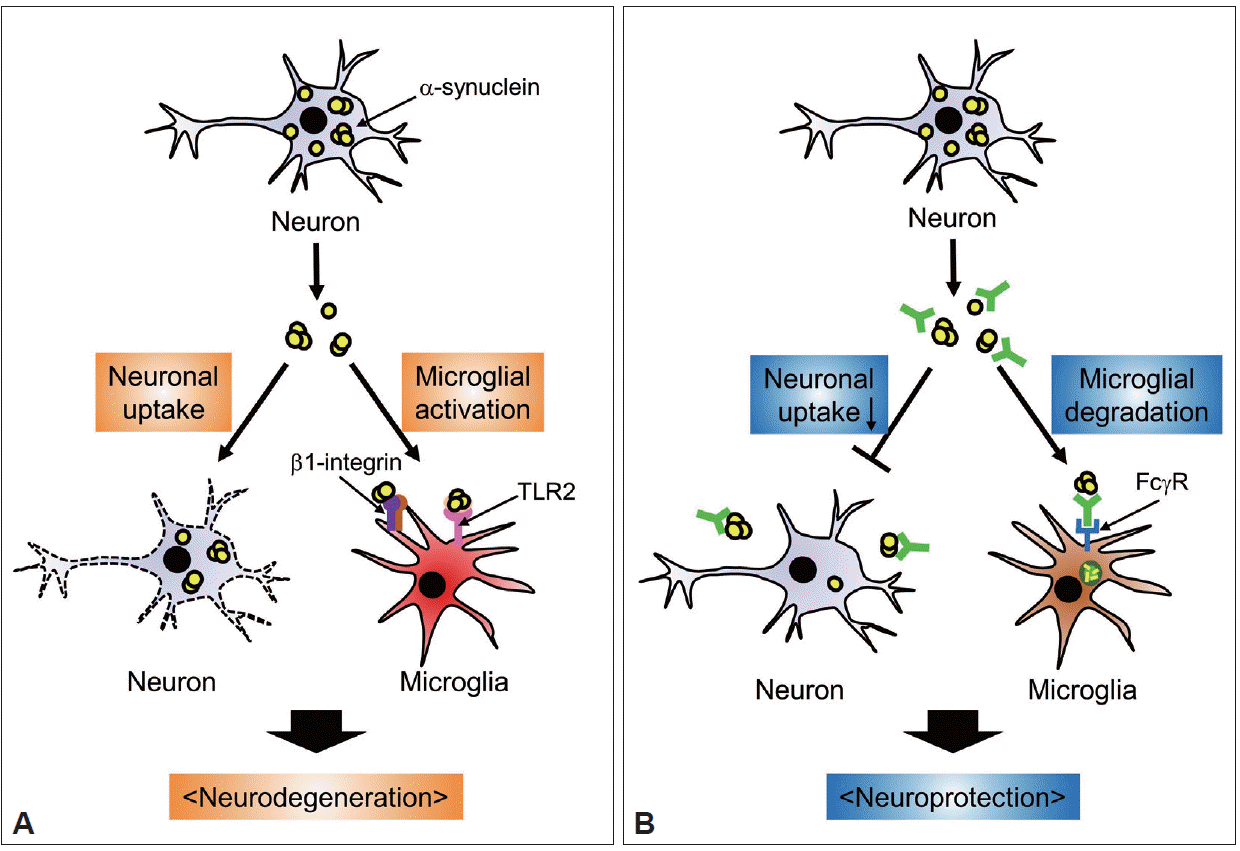
Proposed mechanisms of anti-α-synuclein immunotherapy targeting extracellular α-synuclein. A: Pathogenic roles of extracellular α-synuclein. Cellular α-synuclein is released from neuronal cells into the extracellular space. Extracellular α-synuclein aggregates can be taken up by neighboring neurons, where aggregation of the protein is transmitted. In addition, neuron-released α-synuclein can induce microglial activation and migration by stimulating TLR2 and β1-integrin, respectively. These direct (neuron-to-neuron) and indirect (microglia-mediated) functions of extracellular α-synuclein may account for the neurodegeneration observed in synucleinopathies. B: Mechanisms of anti-α-synuclein immunotherapy. Administration of antibodies targeting α-synuclein may have a neuroprotective effect by 1) blocking the direct transfer of extracellular α-synuclein into neurons and 2) facilitating Fcγ receptor-mediated internalization of extracellular α-synuclein into microglia for subsequent lysosomal degradation. Accelerated clearance would prevent the pathogenic actions of extracellular α-synuclein on neurons and microglia. TLR2: toll-like receptor 2.
PERSPECTIVES
Studies so far have been encouraging in terms of targeting α-synuclein for PD immunotherapy. However, we are facing a number of potential issues or problems in developing immunotherapy for PD. First, immunotherapy should not interfere with the physiological function of α-synuclein. Generation of conformation-specific antibodies for the “pathogenic” forms of α-synuclein would allow us to overcome this possible problem. Second, delivery of antibodies to the brain parenchyma might be a problem. Engineering the antibodies so they can penetrate the blood-brain barrier might be a necessary step. Finally, the efficacy of immunotherapy needs to be validated in non-human primate models. For passive immunization, the antibodies need to be “humanized”, and thus, the toxicity and efficacy of the humanized antibodies have to be tested in non-human primates.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C0093).