 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 8(1); 2015 > Article
-
Review Article
Neurodegeneration with Brain Iron Accumulation: Diagnosis and Management - Penelope Hogarth
-
Journal of Movement Disorders 2015;8(1):1-13.
DOI: https://doi.org/10.14802/jmd.14034
Published online: January 13, 2015
Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, OR, USA
- Corresponding author: Penelope Hogarth, MD, Department of Molecular and Medical Genetics, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Mail Code L-103, Portland, OR 97123, USA, Tel: +1-503-494-7703, Fax: +1-503-494-6886, E-mail: hogarthp@ohsu.edu
• Received: October 13, 2014 • Revised: October 15, 2014 • Accepted: October 20, 2014
Copyright © 2015 The Korean Movement Disorder Society
- ABSTRACT
- INTRODUCTION
- PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION (PKAN)
- PHOSPHOLIPASE A2-ASSOCIATED NEURODEGENERATION (PLAN)
- MITOCHONDRIAL MEMBRANE PROTEIN-ASSOCIATED NEURODEGENERATION (MPAN)
- BETA-PROPELLER PROTEIN-ASSOCIATED NEURODEGENERATION (BPAN)
- OTHER FORMS OF NBIA
- CONCLUSIONS
- Acknowledgments
- Acknowledgments
- Notes
- REFERENCES
ABSTRACT
- Neurodegeneration with brain iron accumulation (NBIA) encompasses a group of inherited disorders that share the clinical features of an extrapyramidal movement disorder accompanied by varying degrees of intellectual disability and abnormal iron deposition in the basal ganglia. The genetic basis of ten forms of NBIA is now known. The clinical features of NBIA range from rapid global neurodevelopmental regression in infancy to mild parkinsonism with minimal cognitive impairment in adulthood, with wide variation seen between and within the specific NBIA sub-type. This review describes the clinical presentations, imaging findings, pathologic features, and treatment considerations for this heterogeneous group of disorders.
- Neurodegeneration with brain iron accumulation (NBIA) comprises a heterogeneous group of inherited neurodegenerative disorders collectively characterized by extrapyramidal movement disorders and abnormal iron accumulation in the deep basal ganglia nuclei of the brain. Ten NBIA genes have been identified to date: eight are autosomal recessive, one is autosomal dominant, and one is X-linked dominant (Table 1). Prevalence data is incomplete, but all forms of NBIA are considered to be “ultra-rare” with less than 1/1000000 affected. Four types of NBIA predominate as shown in Figure 1: pantothenate kinase-associated neurodegeneration (PKAN); phospholipase A2-associated neurodegeneration (PLAN); mitochondrial membrane protein-associated neurodegeneration (MPAN); and beta-propeller protein-associated neurodegeneration (BPAN). This distribution may shift slightly over time as new cases of more recently discovered mutations such as WDR45 grow in number.
INTRODUCTION
- Most of the cases reported in the historical literature as “Hallervorden-Spatz disease” were probably PKAN, but almost certainly included other forms of NBIA, an important observation when trying to glean insights about PKAN from the older literature. Julius Hallervoden and Hugo Spatz were German neuropathologists whose work derived from pathological samples obtained under the Nazi program of active euthanasia of individuals with physical and intellectual disabilities. The PANK2 gene discovery [1] provided not only an opportunity to establish a naming convention for NBIA, but also the catalyst to dishonor and abandon the eponym [2,3]. PKAN represents the most prevalent form of NBIA, accounting for half of the patients in our research registry (Figure 1). The PANK2 gene encodes pantothenate kinase 2, which phosphorylates vitamin B5, N-pantothenoyl cysteine, and pantetheine in the first key regulatory step of coenzyme A biosynthesis, and thus is essential for intermediary and fatty acid metabolism.
- Pantothenate kinase-associated neurodegeneration has traditionally been divided into early-onset, rapidly progressive “classic” disease and a more slowly-progressive “atypical” form that has a later onset and slower progression [4], though there certainly are cases that fall somewhere between, combining features of both forms. In classic PKAN, the child presents before the age of six, usually with a change in gait and falls, sometimes superimposed on a history of mild developmental delay. The neurologic exam at presentation generally reveals evidence of dystonia, with the lower limbs predominantly affected: a striatal toe sign may appear intermittently during the exam, or there may be dystonic posturing of the child’s foot during gait testing. Corticospinal tract signs may also be present, with spasticity, brisk reflexes and extensor plantar responses seen. There may be a history of poor night vision, and formal ophthalmologic evaluation often reveals pigmentary retinopathy with an abnormal electroretinogram, bilateral Adie’s pupil, and various abnormalities of pursuit and saccadic eye movements [5]. Pathognomonic abnormalities are seen on MRI imaging of the brain, even early in disease (see Imaging, below). Acanthocytes may be seen on prepared blood smears [6].
- The progression of classic PKAN is not linear, but rather stepwise, with periods of rapid decline occurring, usually without clear precipitant. As the trunk becomes involved, the child may exhibit a striking opisthotonic posturing that is a hallmark of classic PKAN. Limb dystonia may be curiously asymmetric despite the symmetry of the pallidal lesions on MRI (Figure 2) but impairment of gait and balance is universal and results in most children needing a wheelchair before adolescence. Speech and swallowing are affected as the disease involves the bulbar musculature, causing nutritional compromise and presenting a risk of aspiration pneumonia. Some children with classic PKAN succumb to complications of their disease in the first decade, but many survive into adulthood, albeit with severe disability.
- When PKAN presents later in life, signs and symptoms are far more heterogeneous. The teenager or young adult with atypical PKAN may present with a change in speech patterns, manifesting as stuttering, a Parkinsonian-type palilalia or hypophonia, spasmodic dysphonia, or dysarthria due to oropharyngeal dystonia. A new tic disorder may appear, or there may be a marked worsening of tics attributed earlier in childhood to Tourette syndrome. Neuropsychiatric symptoms are also common in atypical PKAN. Mood lability, impulsivity, non-specific behavioral changes, and obsessive-compulsive features may be early signs and may be attributed to typical changes of adolescence until other neurologic signs appear, prompting further workup. Typical motor signs of illness may not be apparent until later in disease, with most patients exhibiting mixed dystonia and parkinsonism and varying degrees of spasticity. The extrapyramidal motor symptoms in atypical PKAN show an age dependency, with adolescents demonstrating more dystonia than parkinsonism, while the opposite pattern is observed in patients presenting in their 20s, who show bradykinesia, rigidity, freezing of gait and postural instability on exam; a rest tremor is less commonly a feature. Action-induced dystonias are frequently seen in atypical PKAN and may offer an important clue to diagnosis. Oromandibular dystonia triggered by eating and speaking is particularly unusual among the secondary dystonias, chorea-acanthocytosis, the related McLeod acanthocytosis syndrome, and neuroferritinopathy being the only other disorders where eating dystonia is a prominent feature. Writer’s cramp and action-induced dystonic tremors may also be observed. Pigmentary retinopathy is rare in atypical PKAN, though extra-ocular movement abnormalities are common.
- The progression of atypical PKAN is much slower than classic disease, and many individuals have a normal lifespan. Our own observation is that the rate of decline tends to be steeper following the onset of symptoms, but then tends to stabilize and change only minimally over years.
- Cognition may be abnormal in PKAN, but intellectual disability is not a universal part of the disease as suggested by the early “Hallervorden-Spatz” literature and cognitive decline over time is not typical. The younger the onset of disease, the greater the degree of cognitive impairment [7]; this may be because the disease process in very early-onset PKAN disrupts normal brain development. The severity of symptoms may also impact performance on standardized cognitive tests. This, in turn, may lead to overestimation of intellectual disability; a study of children with PKAN undergoing deep brain stimulation surgery found post-surgery improvements in cognitive test performance concomitant with improvement in dystonia symptoms [8].
- Imaging findings in PKAN
- MRI imaging in PKAN is distinctive enough to be diagnostic in most cases. T2 weighted sequences through the basal ganglia reveal the globus pallidus to be hypointense with an anteromedially-placed region of hyperintensity, the so-called “eye of the tiger” sign (Figure 2). The substantia nigra shows normal to mildly hypointense signal. Although these findings in an individual with characteristic signs on exam are highly predictive of the presence of PANK2 mutations, the finding is not absolutely sensitive or specific: rare mutation-positive cases are described in which there is no “eye of the tiger” on MRI [9,10]. Conversely, “eye of the tiger” mimics may be seen in MPAN, carbon monoxide poisoning survivors, multiple system atrophy, neuroferritinopathy, and other unknown diagnoses [11,12].
- Pathology of PKAN
- The pathology of PKAN is much more circumscribed than previously thought, being largely limited to the globus pallidus, in contrast to other forms of NBIA [13]. In gross section, the presence of iron is evidenced by a frankly rusty discoloration of the globus pallidus but not other structures; this is confirmed microscopically with iron-specific stains that reveal the iron to have a perivascular distribution. A central area of neuronal depletion and tissue rarefaction corresponds to the “eye of the tiger” seen on MRI. Two populations of ubiquitin-positive spheroids are seen. Larger pleiomorphic structures thought to represent degenerating “ghost” neurons are concentrated in the globus pallidus but are also noted in the putamen and internal capsule. Smaller eosinophil structures represent the classic neuroaxonal spheroid seen in various forms of NBIA; these are sparsely located in the globus pallidus, corpus callosum and subcortical white matter and stain for amyloid precursor protein as well as ubiquitin.
- Treatment considerations in PKAN
- Currently, treatment is symptomatic. Dystonia and spasticity are usually managed with anticholinergics, benzodiazepines and other anti-spasticity agents such as baclofen, which may be delivered intrathecally. Botulinum toxin injections can also provide targeted relief of dystonia and spasticity. Deep brain stimulation has shown promise, but studies are limited to individual case reports, small case series [14] and a retrospective study [15] which included non-PKAN cases, challenging the generalizability of the results. The ongoing involvement of physical, occupational and speech therapists can delay complications of disease.
- One of the most challenging problems for the patient, family and clinician in PKAN is dystonic crisis or “dystonic storm”. This severe exacerbation of dystonia is seen mostly in children with classic disease and can be life-threatening. It can occur without an obvious precipitant, but the child should be screened for infection, fecal impaction, and occult fractures to be certain there is not a treatable cause. The torsional stress created by the severe dystonia of classic PKAN can result in occult fractures of long bones, especially in children who are no longer weight-bearing and may be osteopenic. The treatment of dystonic storm is very challenging; no controlled studies have been published but treatment strategies have been reviewed in two publications [16,17].
- The contribution of iron accumulation to the core pathophysiology of the disease remains in question [18], but until that question is answered, iron chelation remains under investigation as a disease-modifying approach. A preliminary study of deferiprone, an iron chelator that readily crosses the blood-brain barrier, showed robust reduction of brain iron on brain MRI in PKAN patients, but no measurable benefit in clinical disease outcomes [19]. A double-blind, placebo-controlled international multicenter clinical trial is underway to more rigorously investigate the drug’s benefit (https://clinicaltrials.gov/ct2/show/NCT01741532?term=deferiprone+PKAN&rank=1). A rational therapeutic approach aimed at bypassing the enzymatic defect with a novel compound is also under investigation.
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION (PKAN)
- Mutations in the calcium-independent phospholipase A2 gene PLA2G6, thought to play a critical role in cell membrane phospholipid homeostasis, are responsible for PLAN. When PLAN presents in early childhood, it is called infantile neuroaxonal dystrophy (INAD), a term that originates from the hallmark pathologic finding of dystrophic axons found on nerve or conjunctival biopsy; before genetic testing was available, this finding confirmed the diagnosis. While we have tried to rationalize the rules surrounding nomenclature in the NBIA field by naming each disease using the “(mutant protein)-associated neurodegeneration” convention, INAD is firmly established in the literature and continues to be routinely used for the infantile-onset form of PLAN.
- Classic INAD is a devastating syndrome of neurodevelopmental regression. It presents between 6 months and 3 years of age, initially with slowing or cessation of development, followed by progressive loss of previously acquired milestones in all domains. Truncal hypotonia appears early in the course, sometimes leading to consideration of spinal muscular atrophy in the differential diagnosis. Optic atrophy, strabismus and nystagmus are prominent and are accompanied by visual impairment leading to eventual blindness in many cases. Hypotonia and areflexia is replaced by spastic tetraplegia as the disease advances, a result of early peripheral denervation from an axonal sensorimotor neuropathy and later pyramidal dysfunction. Electroencephalography is abnormal with frontal-predominant fast rhythms and sometimes overt epileptiform discharges. Though seizures have been described as rare and only occurring late [20] a recent review of 25 children with INAD noted epilepsy in 17% [21]. Most children with classic INAD die before the age of 10.
- When the onset of PLAN is later in childhood, and for a minority of patients initially presenting as classic INAD, the disease’s presentation is more variable and its progression slower. These children are said to have “atypical infantile neuroaxonal dystrophy” (aNAD). Their picture differs from the classic form of the disease: cerebellar dysfunction with gait ataxia and dysarthria are more evident, and hypotonia and areflexia tend to predominate, rather than spasticity. Additionally, dystonia may be present. The cerebellar presentation in aNAD may simply be a function of timing of the disease in relation to development: children with classic INAD lose their ability to walk and talk so early in the disease course that signs of cerebellar disease may never be clinically apparent. Cognition is impaired: the child may be initially assessed as having a static encephalopathy, but progressive cognitive decline eventually becomes apparent, sometimes the first clue that the child or teen has a neurodegenerative disorder.
- The PLA2G6 gene discovery and advances in molecular genetic technology led to the recognition of an adult form of PLAN presenting with dystonia-parkinsonism and spasticity, along with cognitive and psychiatric features [22]. Cerebellar features, denervation on electromyography, and fast rhythms on electroencephalography are less common in adult-onset patients than in INAD or aNAD. The parkinsonism is often nicely responsive to dopaminergic medications, but motor fluctuations tend to emerge quickly and complicate management.
- Imaging findings in PLAN
- Cerebellar atrophy is the most common neuroimaging finding in both INAD and aNAD. It is present in the majority of patients even when imaged early in disease, and in virtually 100% of well-established INAD cases. It is an important clue to the diagnosis in a child undergoing workup for psychomotor regression. Additional features seen on MRI include diffuse T2 white matter hyperintensities, and thinning of the corpus callosum and optic chiasma. The imaging of late-onset PLAN is more variable in its features: cerebellar atrophy is inconsistently present though cerebral volume loss may be evident, especially later in the disease. In all forms of PLAN, iron accumulation, manifesting as T2 hypointensity, may be absent or subtle early in the disease course and may never become evident in INAD and aNAD; susceptibility weighted imaging (SWI) may reveal iron not apparent on standard T2 sequences. Iron is generally evident in the globus pallidus on MRI imaging in late-onset patients presenting with clear-cut motor signs of dystonia and parkinsonism (Figure 2).
- Pathology of PLAN
- Pathologic studies are limited to case reports and small case series. Consistent with the MRI findings during life, cerebellar and cortical atrophy may be observed on gross pathology, with widespread neuronal loss and gliosis evident microscopically. Dystrophic axonal spheroids, historically the hallmark diagnostic finding, are seen as eosinophilc swellings in peripheral nerves, spinal cord, brainstem and basal ganglia. A more recently recognized pathologic feature of PLAN is the prominent Lewy body pathology involving the basal ganglia and neocortex, particularly striking in disease of later onset and longer duration. Tau pathology may also be observed with hyperphosphorylated neurofibrillary tangles and neuropil threads [23,24].
- Treatment considerations in PLAN
- Like other forms of NBIA, treatment of PLAN is geared towards relief of symptoms and prevention of complications. Standard medications to treat seizures, spasticity, dystonia, and parkinsonism are employed, though treatment with levodopa of adult-onset dystonia-parkinsonism is complicated by early motor fluctuations and exacerbation of neuropsychiatric symptoms. Physiotherapy early in INAD/aNAD may delay or prevent contractures. Gastrostomy tube placement supports nutritional status as the disease advances.
- Prospects for treatment are perhaps more immediate in PLAN than in some other forms of NBIA, in part because mouse models recapitulate important aspects of the disease, providing a mechanism to test rational therapeutics. Although no clinical trials are yet underway, mouse studies have revealed reduced incorporation of docosahexanoic acid into the mutant mouse brain using novel imaging techniques [25–27], as well as derangements in calcium signaling [28], suggesting future directions for both imaging biomarker development and therapeutics. Perhaps most encouraging, a “proof of concept” study testing a gene therapy approach is underway in the mouse model [29].
PHOSPHOLIPASE A2-ASSOCIATED NEURODEGENERATION (PLAN)
- Mitochondrial membrane protein-associated neurodegeneration is caused by mutations in c19orf12. The protein’s function is poorly understood, but preliminary functional experiments and its sub-cellular association with the mitochondrial membrane suggest a role in cellular energetics and fatty acid metabolism.
- Clinical findings in MPAN
- Mitochondrial membrane protein-associated neurodegeneration typically presents in the first decade of life, but may also have its onset in early adulthood. In childhood, development of a spastic gait with extensor plantar responses is typically the earliest sign, commonly accompanied by optic atrophy, learning difficulties, dysarthria, and sometimes behavioral and psychiatric features. Dystonia, when present, tends to be limited to the feet and hands. The initial presentation in adulthood is more variable, but typically manifests with cognitive and behavioral changes, parkinsonism and mixed gait disorders [30,31].
- Generally, the disease progresses slowly, and most individuals with childhood onset survive into their 20s or beyond. However, some individuals may exhibit rapid and terminal progression, either immediately following the onset of disease or after a period of slow decline. Interestingly, the literature and our own experience suggest that this occurs primarily in adult-onset patients [31,32]. This is in contrast to PKAN and PLAN, where rapid progression is associated with childhood-onset disease.
- As the disease progresses, lower motor neuron signs may emerge, particularly in childhood-onset patients. Clinically, this manifests as loss of deep tendon reflexes, muscle weakness and sometimes atrophy; electrophysiologically, as a motor neuronopathy/axonopathy [31,33–35]. The combination of upper and lower motor neuron findings may raise the question of amyotrophic lateral sclerosis [35,36]. Cognitive decline appears to be universal in MPAN, sometimes accompanied by disabling psychiatric features; most patients are overtly demented by mid-stage disease. Bowel and bladder incontinence are common; it is not clear whether this is due to an autonomic neuronopathy or simply a non-specific feature associated with progressive dementia. Dysphagia is also common later in disease; this may lead to aspiration pneumonia.
- Imaging findings in MPAN
- Pallidal and nigral iron accumulation is evident on brain MRI on T2 and GRE sequences, variably accompanied in some patients by hyperintense streaking of the globus pallidus in the region of the medial medullary lamina (Figure 2). This may be interpreted as an “eye of the tiger” leading to an erroneous radiologic diagnosis of PKAN. Cortical and cerebellar atrophy may be seen in more advanced disease, and T1 hyperintensity in the caudate and putamen has been reported [31,35,37].
- Pathology of MPAN
- Pathologically, MPAN is a synucleinopathy, exhibiting a remarkable burden of Lewy bodies and Lewy neurites not only in the basal ganglia but also in the neocortex [31]. Cortical Lewy body pathology in MPAN exceeds that seen in sporadic Parkinson disease by 40-fold. Even Lewy body dementia, the archetypal cortical Lewy body disease, does not rival MPAN in this regard. Axonal spheroids, thought to represent dying neurons, are seen both peripherally and centrally. Iron deposits are seen in the globus pallidus and to a lesser extent in the substantia nigra; little or no iron is seen in the cortex. Neuronal loss and gliosis are especially prominent in the substantia nigra.
- Treatment considerations in MPAN
- Like all NBIAs, treatment is symptomatic. Anti-spasticity agents such as baclofen may be helpful and allow patients to maintain ambulation for a longer period. Trihexyphenidyl may provide relief when dystonia is problematic. Even when parkinsonism dominates the motor dysfunction, dopaminergic compounds seem to offer only modest relief at best, and may precipitate adverse neuropsychiatric effects; dopamine agonists in particular should be used with caution. There are no reports of deep brain stimulation in MPAN, but the prominent cognitive decline in the disease would strongly argue against its use. Treatment of bladder incontinence ideally should be guided by urodynamic studies and may need to be reassessed as the disease progresses, particularly if patients develop prominent lower motor neuron signs when urinary retention and overflow incontinence may replace a hyperactive detrusor. Standard bowel regimens may lessen the risks of impaction and fecal incontinence. A gastrostomy tube may maintain nutritional status and prolong life as dysphagia progresses.
- When present, psychiatric features such as emotional lability, agitation, hallucinations, and compulsive behaviors can be particularly challenging for families. The close involvement of a psychiatrist familiar with treating cognitively-impaired patients is ideal. For patients with neuropsychiatric complications and prominent parkinsonism in particular, the choice of antipsychotic agent must be made with care and dosed cautiously.
MITOCHONDRIAL MEMBRANE PROTEIN-ASSOCIATED NEURODEGENERATION (MPAN)
- Beta-propeller protein-associated neurodegeneration is unique among the NBIAs in its mode of inheritance, its presumed pathophysiology, its biphasic clinical profile, and its distinctive imaging characteristics. The only X-linked form of NBIA to date and a rare example of X-linked dominant inheritance, BPAN appears to have two distinct phases to its course, comprising a syndrome of global development delay with seizures, pyramidal signs and disordered sleep in childhood, followed by a decline in adulthood with the development of parkinsonism, dystonia and dementia [38]. Prior to the discovery of the causative gene, BPAN was described as “static encephalopathy with neurodegeneration in childhood” (SENDA), but it has now been named according to the established naming convention. The disease is caused by mutations in WDR45, which encode for a beta-propeller protein with a presumed critical function in autophagy based on the yeast homolog and preliminary functional work in humans [39,40]. Although dysregulation of autophagy has been proposed as having a role in neurodegeneration, BPAN is the first disorder in which a direct connection has been made.
- The X-linked dominant pattern of inheritance means that most patients with BPAN will be female. The majority of BPAN cases arise as a result of de novo mutations early in development. In theory, a female with BPAN could arise as the result of either germline or somatic mutations; however, the severity of the phenotype means that most girls with BPAN will never reproduce. Skewing of X-inactivation could account for a mild phenotypes in females, though, and such cases are beginning to be reported [41]. Because affected males have only one copy of the X chromosome, the disease would be predicted to be male-lethal unless the mutation occurs post-zygotically; however, a recent report describes germline mutations in monozygotic twins and their older brother, all of whom exhibited intellectual disability and seizures [42]. The boys’ mother harbored the same mutation but was reported to be phenotypically normal, suggesting that perhaps the transmitted mutation had only minimal effects on function, allowing male survival despite hemizygosity. Clearly, the BPAN story is still unfolding with more surprises to come.
- Clinical features of BPAN
- The child with BPAN may initially appear normal, but it quickly becomes apparent that developmental milestones are not being attained on schedule. Motor milestones are delayed: the child may exhibit toe walking or a broad-based, ataxic gait. Intellectual disability is marked: little expressive language is acquired, with most children attaining only a few spoken words. Seizures are common, and may represent a major source of morbidity for the child with BPAN. On one end of the spectrum is the child who has only one or two seizures during childhood, none at all, or only febrile seizures; these children may require no treatment with anti-epileptic drugs. Other children, however, exhibit a picture of a severe syndromic epilepsy, with multiple seizure types present (partial complex, generalized tonic-clonic, atonic and/or myoclonic) that may be refractory to therapy. Some children exhibit sleep disorders, including hypersomnolence, hyposomnolence, shortened sleep latency, and abnormal rapid eye movement sleep. The presence of hand stereotypies in some children with BPAN, along with marked abnormalities in language development, disordered sleep, and seizures may have earned them a diagnosis of “atypical Rett syndrome” before mutations in WDR45 are found.
- Children with BPAN make slow developmental gains in childhood, though remaining far behind their developmentally-normal peers. During the teenage years or early adulthood, signs of parkinsonism emerge, sometimes fairly precipitously. The posture becomes stooped, movements slow, and the gait changes, with shortened step length and freezing. While these symptoms are responsive to dopaminergic medications, sometimes robustly so, the benefit tends to be short-lived, with brittle levodopa-induced dyskinesias or dystonia quickly complicating management. In addition to the motor signs, a progressive decline in cognitive function becomes evident and previously-learned skills are lost. Challenging new behaviors may appear, often exacerbated by dopaminergic drugs. Though the decline may be gradual, eventually all patients lose ambulatory ability and become profoundly demented.
- Imaging of BPAN
- Imaging in early childhood is typically unremarkable. Later in the course, particularly as parkinsonism becomes evident on exam and prompts further diagnostic studies, a characteristic pattern appears on MR imaging. On T2 sequences, the globus pallidus, substantia nigra and cerebral peduncles become hypointense consistent with iron accumulation in these structures; these findings are more apparent and seen earlier in the disease with the use of iron-sensitive sequences such as T2* and susceptibility-weighted imaging. The hypointensity is most pronounced in the substantia nigra where it appears as a discrete linear streak. This same area on T1 sequences is surrounded by a hyperintense “halo” extending to the cerebral peduncles, thought to represent neuromelanin release from degenerating neurons (Figure 2) [38,43,44]. Derangements in white matter architecture and impaired pallidal and nigral metabolism have been demonstrated in a single adult BPAN case using diffusion tensor imaging and MR spectroscopy [44]. Other findings that may be seen on MRI include thinning of the corpus callosum, cerebellar atrophy and more global atrophy as the disease advances [38,40].
- Pathology of BPAN
- Grossly, cerebellar atrophy and thinning of the cerebral penduncles is apparent [38]. The iron accumulation can be seen in the unstained brain as a rusty discoloration in the substantia nigra. The iron is much less prominent in the unstained globus pallidus, but both structures show strong reaction when treated with iron-specific stains such as Prussian blue. Despite the prominent parkinsonism seen clinically, BPAN does not demonstrate the α-synuclein pathology seen in idiopathic Parkinson disease; rather, tau positive neurofibrillary tangles are seen in the cortex, putamen, hippocampus and hypothalamus. Axonal spheroids, a pathologic feature representing dying neurons and common to many forms of NBIA, are readily seen in the substantia nigra and globus pallidus, as well as in the medulla, pons and thalamus.
- Treatment considerations in BPAN
- As the most recent form of NBIA to be characterized, we are only just starting to understand the phenotypic spectrum of BPAN and define optimal approaches to management. In childhood, the most challenging problem is refractory seizures. Although only present in a minority, these BPAN patients should have the close involvement of a pediatric specialist experienced in the management of severe syndromic epilepsies. In adulthood, the parkinsonism can be treated successfully with dopaminergic medications, although as mentioned, motor fluctuations and dyskinesias pose problems and the drug benefit is not durable. Dopamine agonists might be predicted to have adverse neuropsychiatric effects in BPAN where cognitive impairment is a prominent part of the phenotype [45]; however, we have not seen that in our own clinic.
- Preliminary work to create a mouse model of BPAN is yielding promising results [46]. As we begin to understand the role of autophagy in the disorder, we anticipate the development of rational therapeutics targeting the underlying defect.
- Our own observations of the phenotype in BPAN to date are based on a highly homogeneous cohort selected specifically for gene discovery efforts. As more patients are found earlier in childhood through whole exome sequencing, we expect to see greater phenotypic variability, particularly since timing of somatic mutation and X-inactivation patterns likely influence the phenotype.
BETA-PROPELLER PROTEIN-ASSOCIATED NEURODEGENERATION (BPAN)
- Fatty acid hydroxylase-associated neurodegeneration (FAHN)
- The FA2H gene product is responsible for hydroxylating fatty acids and plays a key role in myelin production in the central nervous system and possibly in cell cycle regulation [47]. Mutations in the gene were originally described as causing leukodystrophy and a complicated form of hereditary spastic paraplegia (HSP35) [48–50]. Subsequent investigations revealed the presence of pallidal T2 hypointensities in some patients harboring FA2H mutations and led to fatty acid hydroxylase-associated neurodegeneration’s (FAHN’s) designation as a subtype of NBIA [51]. The disease usually presents in the first decade of life with gait difficulties and falling. Progressive spasticity, dystonia and cerebellar dysfunction contribute to disability, leading to loss of ambulation in many cases, along with dysarthria and dysphagia. Optic atrophy is frequently present on exam, with variable degrees of accompanying visual impairment. Most children exhibit progressive cognitive decline; seizures are less consistently a feature. An axonal neuropathy has been described in one family [52]; further study is needed to see if this is a consistent part of the phenotype. In addition to evidence of iron accumulation in the globus pallidus (which is not always present), MR imaging of the brain reveals T2-bright white matter lesions, thinning of the corpus callosum, and progressive atrophy of cerebellum, pons, medulla and cord [53].
- Many of FAHN’s clinical features mirror those of INAD/aNAD; thus, a child suspected of having PLAN but without mutations in PLA2G6 should be tested for FAHN. Like other forms of NBIA, the phenotypic spectrum of the disease is expected to broaden as more patients are discovered through whole exome sequencing.
- COASY protein-associated neurodegeneration (CoPAN)
- COASY protein-associated neurodegeneration (CoPAN) joins PKAN as the second inborn error of coenzyme A metabolism. CoPAN manifests in the first decade of life with gait difficulties and mild cognitive impairment. Oromandibular dystonia, dysarthria, and progressive spasticity follow, along with the appearance of an axonal neuropathy. The emergence of parkinsonism further adds to the disability. MRI demonstrates non-homogenous T2 pallidal hypointensity with a region of medial hyperintensity that is reminiscent of the “eye of the tiger” sign seen in PKAN. Although only two families with CoPAN have been reported thus far [54], the discovery that mutations in the gene encoding Coenzyme A synthase (COASY) could cause a form of NBIA confirms the importance of the CoA pathway in neuronal health.
- Neuroferritinopathy
- Mutations in the gene encoding ferritin light chain protein cause neuroferritinopathy, a dominantly-inherited syndrome of chorea, dystonia, parkinsonism, cognitive decline and low serum ferritin that typically presents in mid-life. A majority of the cases described in the literature are clustered geographically in the Cumbrian region of Britain and have a common 460insA mutation [55], suggesting a founder effect. Single families with private mutations and slightly different phenotypes have been described in Europe, North America, and Japan. Neuroferritinopathy can be distinguished from Huntington disease by its prominent action-induced orofacial dystonia, asymmetric presentation, late cognitive decline and by the presence of abnormal iron accumulation on brain MRI in the caudate, putamen, thalamus, globus pallidus, substantia nigra and red nucleus. Cystic change of the caudate and putamen occurs late in the disease [56,57]. Neuroferritinopathy differs from other forms of NBIA by its dominant inheritance pattern, uncommon presentation in childhood, and distinctive pattern of iron accumulation. Like aceruloplasminemia but distinct from most forms of NBIA, the pathophysiology of neuroferritinopathy explains the brain iron accumulation: the mutant protein disrupts the structure and iron-carrying capacity of ferritin, resulting in abnormal iron deposition in the brain.
- Aceruloplasminemia
- Aceruloplasminemia presents in adulthood and is characterized by microcytic anemia, diabetes, retinal disease, and a movement disorder consisting of facial dystonia, chorea, tremor, parkinsonism, ataxia, and cognitive decline [58,59]. In addition to anemia, laboratory investigations reveal low or absent serum ceruloplasmin, elevated ferritin, low iron, and low serum copper but normal urinary copper. Like neuroferritinopathy, it is a disorder of iron metabolism: mutations in the CP gene cause an absence of or reduction in the copper-carrying ceruloplasmin protein. This results in abnormal iron trafficking and deposition throughout the body, including the central nervous system. Iron chelation is routinely employed as a rational therapeutic, but while changes in iron deposition in peripheral organs and the brain can be demonstrated after several years of treatment, it is not clear whether this is accompanied by significant clinical improvement [60,61].
- Woodhouse-Sakati syndrome
- DCAF17 encodes a nucleolar protein thought to be involved in transcriptional regulation, and mutations in the gene result in an unusual syndrome of endocrine and neurologic abnormalities [62]. Affected individuals exhibit hypogonadism, diabetes mellitus, alopecia, abnormalities on electrocardiogram, extrapyramidal movement disorders, intellectual disability, and sensorineural healing loss [63,64]. MRI imaging reveals basal ganglia T2 hypointensities as well as white matter disease. Most of the described cases arise from a founder in the Saudi Arabian population, though affecteds of other ethnicities have been described [65].
- Kufor-Rakeb syndrome
- Kufor-Rakeb syndrome is a syndrome of juvenile-onset parkinsonism, spasticity, and cognitive decline originally described in a Jordanian family [66] and now described in multiple other ethnicities [67–71]. Variable clinical features include supranuclear gaze palsy, facial-finger-faucial mini myoclonus, and tremor. Generalized atrophy is a universal finding on brain MRI as the disease advances; basal ganglia iron may not be evident, especially early in disease. The parkinsonism is levodopa-responsive but, like MPAN and BPAN, management is complicated by the early development of motor fluctuations and dyskinesias. The disease is caused by mutations in the APT13A2 gene [72 encoding a lysosomal ATPase and is also known as PARK9.
OTHER FORMS OF NBIA
- The development of animal and cell-based model systems are advancing our understanding of the core pathophysiology of the disorders of brain iron accumulation, individually and collectively. Patient registries, natural history studies and well-curated biorepositories will be essential to support drug development efforts as rational therapeutic approaches to disease modification arise from the laboratory. Such efforts are underway.
CONCLUSIONS
- We gratefully acknowledge the participation of NBIA patients and families in our research, and the support of or our work by the NBIA Alliance including the NBIA Disorders Association and Hoffnungsbaum e.V.
- This publication was supported by Oregon Clinical and Translational Research Institute (OCTRI), grant number (UL1TR000128) from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Hogarth also receives funding support for NBIA research from the NBIA Disorders Association, the European Commission’s 7th Framework Programme (FP7/2007–2013, HEALTH-F2-2011, grant agreement No. 277984, TIRCON), and Retrophin, Inc. She receives funding support for work unrelated to NBIA from the Michael J. Fox Foundation, and Vertex, Inc.
Acknowledgments
Acknowledgments
Figure 1Distribution of NBIA subtypes in the North American database. NBIA: neurodegeneration with brain iron accumulation, PKAN: pantothenate kinase-associated neurodegeneration, PLAN: phospholipase A2-associated neurodegeneration, INAD: infantile neuroaxonal dystrophy, MPAN: mitochondrial membrane protein-associated neurodegeneration, BPAN: beta-propeller protein-associated neurodegeneration, FAHN: fatty acid hydroxylase-associated neurodegeneration, CoPAN: Coenzyme A synthase protein-associated neurodegeneration, NF: neuroferritinopathy, KRS: Kufor-Rakeb syndrome, ACP: aceruloplasminemia.
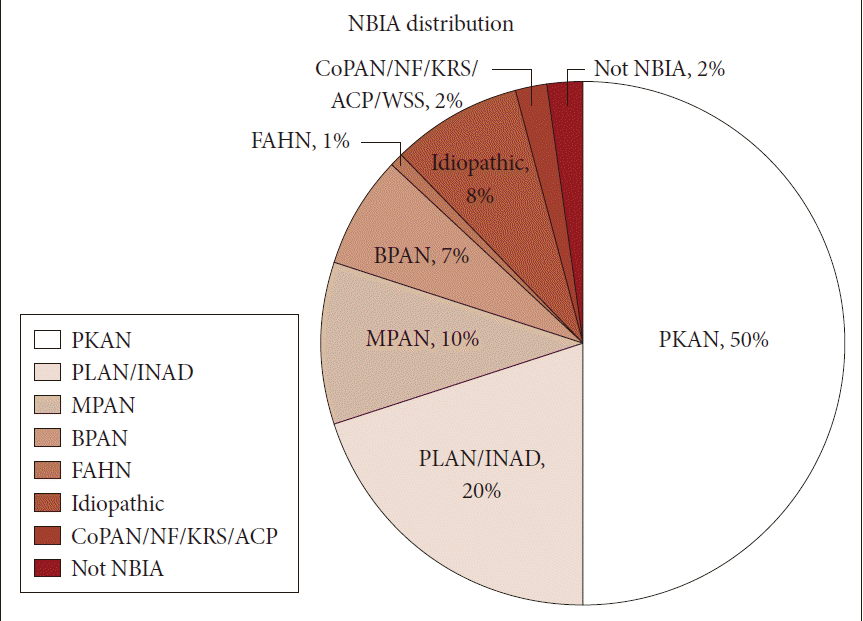
Figure 2Imaging characteristics of the four major subtypes of NBIA. All images performed on 3.0T magnet except (G) and (H) which were performed on 1.5T. A: T2-weighted imaging in PKAN shows GP hypointensity indicating iron accumulation with an anteromedially-located area of hyperintensity, the so-called “eye of the tiger”. B: SN in same patient showing hypointensity in medial aspect of nucleus. C: T2-weighted sequence in a patient with MPAN showing pallidal hypointensity with hyperintense streaking in the region of the medial medullary lamina. Depending on the cut, this may be mistaken for an “eye of the tiger” characteristic of PKAN. D: The SN in the same patient also demonstrates iron accumulation. E: T2 sequence of GP and (F) SN in a 9 yo child with PLAN showing evidence of iron accumulation. Imaging performed earlier in the disease course had shown no signal changes. Inset in (F) showing cerebellar atrophy in the same child. G: T2 imaging showing the GP in a young adult with BPAN, after the onset of parkinsonian symptoms. In (H) note the marked hypointensity in the SN, and, in the inset in (H), the same region on T1 weighted sequence showing the characteristic hyperintense “halo” thought to represent neuromelanin release from degenerating neurons. PKAN: pantothenate kinase associated neurodegeneration, MPAN: mitochondrial membrane protein-associated neurodegeneration, BPAN: beta-propeller protein-associated neurodegeneration, PLAN: phospholipase A2-associated neurodegeneration, GP: globus pallidus, SN: substantia nigra, NBIA: neurodegeneration with brain iron accumulation.
Table 1The ten forms of NBIA described to date, with name, acronym, mutated gene, and mode of inheritance
- 1. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 2001;28:345–349.ArticlePubMedPDF
- 2. Shevell M. Hallervorden and history. N Engl J Med 2003;348:3–4.ArticlePubMed
- 3. Zeidman LA, Pandey DK. Declining use of the Hallervorden-Spatz disease eponym in the last two decades. J Child Neurol 2012;27:1310–1315.ArticlePubMed
- 4. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003;348:33–40.ArticlePubMed
- 5. Egan RA, Weleber RG, Hogarth P, Gregory A, Coryell J, Westaway SK, et al. Neuro-ophthalmologic and electroretinographic findings in pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). Am J Ophthalmol 2005;140:267–274.ArticlePubMedPMC
- 6. Gregory A, Hayflick SJ. Pantothenate Kinase-Associated Neurodegeneration. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington; 1993–2014.
- 7. Freeman K, Gregory A, Turner A, Blasco P, Hogarth P, Hayflick S. Intellectual and adaptive behaviour functioning in pantothenate kinase-associated neurodegeneration. J Intellect Disabil Res 2007;51(Pt. 6):417–426.ArticlePubMedPMC
- 8. Mahoney R, Selway R, Lin JP. Cognitive functioning in children with pantothenate-kinase-associated neurodegeneration undergoing deep brain stimulation. Dev Med Child Neurol 2011;53:275–279.ArticlePubMed
- 9. Chiapparini L, Savoiardo M, D’Arrigo S, Reale C, Zorzi G, Zibordi F, et al. The “eye-of-the-tiger” sign may be absent in the early stages of classic pantothenate kinase associated neurodegeneration. Neuropediatrics 2011;42:159–162.ArticlePubMedPDF
- 10. Delgado RF, Sanchez PR, Speckter H, Then EP, Jimenez R, Oviedo J, et al. Missense PANK2 mutation without “eye of the tiger” sign: MR findings in a large group of patients with pantothenate kinase-associated neurodegeneration (PKAN). J Magn Reson Imaging 2012;35:788–794.ArticlePubMed
- 11. Strecker K, Hesse S, Wegner F, Sabri O, Schwarz J, Schneider JP. Eye of the tiger sign in multiple system atrophy. Eur J Neurol 2007;14:e1–e2.Article
- 12. Chang CL, Lin CM. Eye-of-the-Tiger sign is not Pathognomonic of Pantothenate Kinase-Associated Neurodegeneration in Adult Cases. Brain Behav 2011;1:55–66.ArticlePubMedPMC
- 13. Kruer MC, Hiken M, Gregory A, Malandrini A, Clark D, Hogarth P, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011;134(Pt 4):947–958.ArticlePubMedPMCPDF
- 14. Castelnau P, Cif L, Valente EM, Vayssiere N, Hemm S, Gannau A, et al. Pallidal stimulation improves pantothenate kinase-associated neurodegeneration. Ann Neurol 2005;57:738–741.ArticlePubMed
- 15. Timmermann L, Pauls KA, Wieland K, Jech R, Kurlemann G, Sharma N, et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain 2010;133(Pt 3):701–712.ArticlePubMedPMCPDF
- 16. Fasano A, Ricciardi L, Bentivoglio AR, Canavese C, Zorzi G, Petrovic I, et al. Status dystonicus: predictors of outcome and progression patterns of underlying disease. Mov Disord 2012;27:783–788.ArticlePubMed
- 17. Allen NM, Lin JP, Lynch T, King MD. Status dystonicus: a practice guide. Dev Med Child Neurol 2014;56:105–112.ArticlePubMed
- 18. Hayflick SJ, Hogarth P. As iron goes, so goes disease? Haematologica 2011;96:1571–1572.ArticlePubMedPMC
- 19. Zorzi G, Zibordi F, Chiapparini L, Bertini E, Russo L, Piga A, et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: results of a phase II pilot trial. Mov Disord 2011;26:1756–1759.ArticlePubMed
- 20. Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, et al. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology 1999;52:1472–1478.ArticlePubMed
- 21. Zhang P, Gao Z, Jiang Y, Wang J, Zhang F, Wang S, et al. Follow-up study of 25 Chinese children with PLA2G6-associated neurodegeneration. Eur J Neurol 2013;20:322–330.ArticlePubMed
- 22. Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 2009;65:19–23.ArticlePubMedPMC
- 23. Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 2008;71:1402–1409.ArticlePubMedPMC
- 24. Paisán-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 2012;33:814–823.ArticlePubMedPMC
- 25. Basselin M, Rosa AO, Ramadan E, Cheon Y, Chang L, Chen M, et al. Imaging decreased brain docosahexaenoic acid metabolism and signaling in iPLA(2)β (VIA)-deficient mice. J Lipid Res 2010;51:3166–3173.ArticlePubMedPMC
- 26. Rapoport SI, Ramadan E, Basselin M. Docosahexaenoic acid (DHA) incorporation into the brain from plasma, as an in vivo biomarker of brain DHA metabolism and neurotransmission. Prostaglandins Other Lipid Mediat 2011;96:109–113.ArticlePubMedPMC
- 27. Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, et al. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A(2)-VIA (iPLA(2)β)-knockout mice. Biochim Biophys Acta 2012;1821:1278–1286.ArticlePubMedPMC
- 28. Strokin M, Seburn KL, Cox GA, Martens KA, Reiser G. Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum Mol Genet 2012;21:2807–2814.ArticlePubMedPMC
- 29. NBIAdisorders.org [Internet]; California: NBIA Disorders Association; February/March newsletter 2014. Available from: http://www.nbiadisorders.org/images/newsletters/2014-feb-mar-news.pdf.
- 30. Gregory A, Hartig M, Prokisch H, Kmiec T, Hogarth P, Hayflick SJ. Mitochondrial Membrane Protein-Associated Neurodegeneration. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJ, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington; 1993–2014.
- 31. Hogarth P, Gregory A, Kruer MC, Sanford L, Wagoner W, Natowicz MR, et al. New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013;80:268–275.ArticlePubMedPMC
- 32. Dogu O, Krebs CE, Kaleagasi H, Demirtas Z, Oksuz N, Walker RH, et al. Rapid disease progression in adult-onset mitochondrial membrane protein-associated neurodegeneration. Clin Genet 2013;84:350–355.ArticlePubMed
- 33. Hartig MB, Iuso A, Haack T, Kmiec T, Jurkiewicz E, Heim K, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet 2011;89:543–550.ArticlePubMedPMC
- 34. Goldman JG, Eichenseer SR, Berry-Kravis E, Zimnowodzki S, Gregory A, Hogarth P, et al. Clinical features of neurodegeneration with brain iron accumulation due to a C19orf12 gene mutation. Mov Disord 2013;28:1462–1463.ArticlePubMed
- 35. Schottmann G, Stenzel W, Lützkendorf S, Schuelke M, Knierim E. A novel frameshift mutation of C19ORF12 causes NBIA4 with cerebellar atrophy and manifests with severe peripheral motor axonal neuropathy. Clin Genet 2014;85:290–292.ArticlePubMed
- 36. Deschauer M, Gaul C, Behrmann C, Prokisch H, Zierz S, Haack TB. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J Neurol 2012;259:2434–2439.ArticlePubMed
- 37. Schulte EC, Claussen MC, Jochim A, Haack T, Hartig M, Hempel M, et al. Mitochondrial membrane protein associated neurodegenration: a novel variant of neurodegeneration with brain iron accumulation. Mov Disord 2013;28:224–227.ArticlePubMed
- 38. Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, et al. β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013;136(Pt 6):1708–1717.ArticlePubMedPMCPDF
- 39. Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012;91:1144–1149.ArticlePubMedPMC
- 40. Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013;45:445–449.449e1.ArticlePubMedPDF
- 41. Harik S, Dandu V, Angtuaco E, Hayflick S. Phenotypic Differences in Identical Twins with Mutated WDR45, a Newly Discovered X-Chromosome Gene Mutation Which Causes Neurodegeneration with Brain Iron Accumulation (NBIA) (P03.052). Neurology 2013;80:P03.052.Article
- 42. Dufke A, Grasshoff U, Dufke C, Kalscheuer V, Schroeder C, Beck-Wödl S, et al. NGS based whole X-exome analysis reveals a familial WDR45 missense mutation in 3 males with intellectual disability and brain iron accumulation [ESHG Abstract P08.53s]. Eur J Hum Genet 2014;22(suppl):161.
- 43. Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, Gregory A, et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol 2012;33:407–414.ArticlePubMedPMC
- 44. Kimura Y, Sato N, Sugai K, Maruyama S, Ota M, Kamiya K, et al. MRI, MR spectroscopy, and diffusion tensor imaging findings in patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Brain Dev 2013;35:458–461.ArticlePubMed
- 45. Verhoeven WM, Egger JI, Koolen DA, Yntema H, Olgiati S, Breedveld GJ, et al. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat Disord 2014;20:332–336.ArticlePubMed
- 46. Biagosch CA, Hensler S, Kühn R, Meitinger T, Prokisch H. TALEN-mediated mutagenesis as a tool to generate disease models for diseases caused by dominant de novo mutations. [ESHG Abstract P08.53s]. Eur J Hum Genet 2014;22(suppl):153.
- 47. Alderson NL, Hama H. Fatty acid 2-hydroxylase regulates cAMP-induced cell cycle exit in D6P2T schwannoma cells. J Lipid Res 2009;50:1203–1208.ArticlePubMedPMC
- 48. Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet 2008;83:643–648.ArticlePubMedPMC
- 49. Dick KJ, Al-Mjeni R, Baskir W, Koul R, Simpson MA, Patton MA, et al. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology 2008;71:248–252.ArticlePubMed
- 50. Dick KJ, Eckhardt M, Paisán-Ruiz C, Alshehhi AA, Proukakis C, Sibtain NA, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat 2010;31:E1251–E1260.ArticlePubMed
- 51. Kruer MC, Paisán-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A, et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 2010;68:611–618.ArticlePubMedPMC
- 52. Pierson TM, Simeonov DR, Sincan M, Adams DA, Markello T, Golas G, et al. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur J Hum Genet 2012;20:476–479.ArticlePubMedPMCPDF
- 53. Kruer MC, Gregory A, Hayflick SJ. Fatty Acid Hydroxylase-Associated Neurodegeneration. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington; 1993–2011.
- 54. Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, Pasqualato S, et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 2014;94:11–22.ArticlePubMedPMC
- 55. Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet 2001;28:350–354.ArticlePubMedPDF
- 56. Chinnery PF, Crompton DE, Birchall D, Jackson MJ, Coulthard A, Lombès A, et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 2007;130(Pt 1):110–119.ArticlePubMedPDF
- 57. Chinnery PF. Neuroferritinopathy. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington; 1993–2014.
- 58. Miyajima H, Takahashi Y, Kono S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals 2003;16:205–213.ArticlePubMed
- 59. Miyajima H. Aceruloplasminemia. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington; 1993–2014.Article
- 60. Mariani R, Arosio C, Pelucchi S, Grisoli M, Piga A, Trombini P, et al. Iron chelation therapy in aceruloplasminaemia: study of a patient with a novel missense mutation. Gut 2004;53:756–758.ArticlePubMedPMC
- 61. Pan PL, Tang HH, Chen Q, Song W, Shang HF. Desferrioxamine treatment of aceruloplasminemia: long-term follow-up. Mov Disord 2011;26:2142–2144.ArticlePubMed
- 62. Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet 2008;83:684–691.ArticlePubMedPMC
- 63. Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet 1983;20:216–219.ArticlePubMedPMC
- 64. Al-Semari A, Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am J Med Genet A 2007;143A:149–160.Article
- 65. Koshy G, Danda S, Thomas N, Mathews V, Viswanathan V. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin Dysmorphol 2008;17:57–60.ArticlePubMed
- 66. Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 1994;89:347–352.ArticlePubMed
- 67. Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007;68:1557–1562.ArticlePubMed
- 68. Crosiers D, Ceulemans B, Meeus B, Nuytemans K, Pals P, Van Broeckhoven C, et al. Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat Disord 2011;17:135–138.ArticlePubMed
- 69. Santoro L, Breedveld GJ, Manganelli F, Iodice R, Pisciotta C, Nolano M, et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 2011;12:33–39.ArticlePubMedPMC
- 70. Brüggemann N, Hagenah J, Reetz K, Schmidt A, Kasten M, Buchmann I, et al. Recessively inherited parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Arch Neurol 2010;67:1357–1363.ArticlePubMed
- 71. Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 2010;25:979–984.ArticlePubMed
- 72. Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006;38:1184–1191.ArticlePubMedPDF
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Olfactory status in neurodegeneration with brain iron accumulation disorders
Elahe Amini, Mohammad Rohani, Maryam Jalessi, Zahra Azad, Franco Valzania, Francesco Cavallieri, Mohammad Farhadi, Zeinab Gholibeigian
Neurological Sciences.2024; 45(2): 647. CrossRef - Neurodegeneration with Brain Iron Accumulation Disorders and Retinal Neurovascular Structure
Elahe Amini, Mohammad Rohani, Alfonso Fasano, Zahra Azad, Shahnaz Miri, Seyed Amir Hassan Habibi, Maziar Emamikhah, Reza Mirshahi, Mohammad Taghi Joghataei, Zeinab Gholibeigian, Khalil Ghasemi Falavarjani
Movement Disorders.2024; 39(2): 411. CrossRef - Femur Fractures in 5 Individuals With Pantothenate Kinase-associated Neurodegeneration: The Role of Dystonia and Suggested Management
Laken Behrndt, Allison Gregory, Katrina Wakeman, Alison Freed, Jenny L. Wilson, Robert Spaull, Manju A. Kurian, Santosh Mordekar, James A. Fernandes, Susan J. Hayflick, Penelope Hogarth, Scott Yang
Journal of Pediatric Orthopaedics.2024; 44(1): e61. CrossRef - Transcranial sonography in neurodegeneration with brain iron accumulation disorders
Seyed Amir Hassan Habibi, Sharmin Aghavali, Zahra Azad, Elahe Amini, Masoumeh Falah, Zeinab Gholibeigian, Narges Yazdi, Maziar Emamikhah, Mohammad Rohani
Clinical Neurology and Neurosurgery.2024; 236: 108074. CrossRef - Estimation of Ambulation and Survival in Neurodegeneration with Brain Iron Accumulation Disorders
Elahe Amini, Mohammad Rohani, Anthony E. Lang, Zahra Azad, Seyed Amir Hassan Habibi, Afagh Alavi, Gholamali Shahidi, Maziar Emamikhah, Ahmad Chitsaz
Movement Disorders Clinical Practice.2024; 11(1): 53. CrossRef - Mitochondrial-Membrane-Protein-Associated Neurodegeneration in Longitudinal Magnetic Resonance Imaging Over 11 Years of Follow-Up
Jiyun Lee, Jin Ju Kim, Chul Hyoung Lyoo, Yun Joong Kim
Journal of Clinical Neurology.2024; 20(2): 220. CrossRef - Clinical, neuroimaging and genetic findings in Brazilian patients with neurodegeneration with brain iron accumulation
Rubens Paulo Araújo Salomão, Flávio Moura Rezende Filho, Vanderci Borges, Manju A. Kurian, Henrique Ballalai Ferraz, Guido J. Breedveld, Vincenzo Bonifati, Orlando G. Barsottini, José Luiz Pedroso
Parkinsonism & Related Disorders.2024; 123: 106103. CrossRef - Genetic Targets and Applications of Iron Chelators for Neurodegeneration with Brain Iron Accumulation
Neharika Marupudi, May P. Xiong
ACS Bio & Med Chem Au.2024;[Epub] CrossRef - Missing heritability of Wilson disease: a search for the uncharacterized mutations
Shubhrajit Roy, Sampurna Ghosh, Jharna Ray, Kunal Ray, Mainak Sengupta
Mammalian Genome.2023; 34(1): 1. CrossRef - A Mild Form of Neurodegeneration with Brain Iron Accumulation attributed to Coenzyme A Synthase Mutation
Narges Hashemi, Reza Nejad Shahrokh Abadi, Afagh Alavi, Ali Reza Tavasoli, Mohammad Rohani
Movement Disorders Clinical Practice.2023; 10(2): 331. CrossRef - Neurodegeneration with brain iron accumulation: a case series highlighting phenotypic and genotypic diversity in 20 Indian families
Haseena Sait, Somya Srivastava, Manmohan Pandey, Deepak Ravichandran, Anju Shukla, Kausik Mandal, Deepti Saxena, Arya Shambhavi, Purvi Majethia, Lakshmi Priya Rao, Suvasini Sharma, Shubha R. Phadke, Amita Moirangthem
neurogenetics.2023; 24(2): 113. CrossRef - Status Epilepticus in Coenzyme A Synthase Protein-Associated Neurodegeneration - Expanding the Clinical Phenotype
Valaparambil Karthika Ajit, Prabhu Selvaraj, KP Divya, Bejoy Thomas, Ramshekhar N. Menon, Soumya Sundaram
Indian Journal of Pediatrics.2023; 90(5): 519. CrossRef - Exploring the genetic and genomic connection underlying neurodegeneration with brain iron accumulation and the risk for Parkinson’s disease
Pilar Alvarez Jerez, Jose Luis Alcantud, Lucia de los Reyes-Ramírez, Anni Moore, Clara Ruz, Francisco Vives Montero, Noela Rodriguez-Losada, Prabhjyot Saini, Ziv Gan-Or, Chelsea X. Alvarado, Mary B. Makarious, Kimberley J. Billingsley, Cornelis Blauwendra
npj Parkinson's Disease.2023;[Epub] CrossRef - Substance abuse and neurodegenerative diseases: focus on ferroptosis
Cheng Guo, Lei Chen, Yun Wang
Archives of Toxicology.2023; 97(6): 1519. CrossRef - Clinical, imaging and genetic profile of twenty-four patients with pantothenate kinase-associated neurodegeneration (PKAN)- A single centre study from India
Neeharika Sriram, Vikram V. Holla, Riyanka Kumari, Nitish Kamble, Jitender Saini, Rohan Mahale, Manjunath Netravathi, Hansashree Padmanabha, Vykuntaraju K. Gowda, Rajani Battu, Akhilesh Pandey, Ravi Yadav, Babylakshmi Muthusamy, Pramod Kumar Pal
Parkinsonism & Related Disorders.2023; 111: 105409. CrossRef - Heme: The Lord of the Iron Ring
Vanessa Azevedo Voltarelli, Rodrigo W. Alves de Souza, Kenji Miyauchi, Carl J. Hauser, Leo Edmond Otterbein
Antioxidants.2023; 12(5): 1074. CrossRef - Beta-propeller protein-associated neurodegeneration: A clinical update with a case report
Moustafa A. Mansour, Yehia Moawad, Hassan Ali
eNeurologicalSci.2023; 31: 100469. CrossRef - The Effects of Modulators of the Coenzyme A Biosynthesis System on Metabolic Stress and the Glutathione System in the CNS in Aluminum Neurotoxicosis
D. S. Semenovich, V. A. Gurinovich, E. P. Lukienko, I. N. Katkovskaya, O. V. Titko, N. P. Kanunnikova, A. G. Moiseenok
Neurochemical Journal.2023; 17(1): 65. CrossRef - Late‐Onset Beta‐Propeller Protein‐Associated Neurodegeneration: A Case Report
Roger Collet‐Vidiella, Gonzalo Olmedo‐Saura, Iñigo Ruiz‐Barrio, Ana Martínez‐Viguera, Benjamin Rodriguez‐Santiago, Sara Bernal, Jaime Kulisevsky, Javier Pagonabarraga
Movement Disorders Clinical Practice.2023; 10(8): 1211. CrossRef - The first reports of FA2H-associated neurodegeneration from two unrelated Iranian families
Narges Hashemi, Reza Nejad Shahrokh Abadi, Afagh Alavi, Mohammad Rohani, Aida Ghasemi, Ali Reza Tavasoli
Neurological Sciences.2023; 44(12): 4359. CrossRef - Autosomal Dominant MPAN: Mosaicism Expands the Clinical Spectrum to Atypical Late‐Onset Phenotypes
Chloé Angelini, Christelle Marie Durand, Patricia Fergelot, Julie Deforges, Anne Vital, Patrice Menegon, Elizabeth Sarrazin, Rémi Bellance, Stéphane Mathis, Victoria Gonzalez, Mathilde Renaud, Solène Frismand, Emmanuelle Schmitt, Marie Rouanet, Lydie Burg
Movement Disorders.2023; 38(11): 2103. CrossRef - Treatment of Pantothenate-Kinase Neurodegeneration With Baclofen, Botulinum Toxin, and Deferiprone: A Case Report
Marya Hameed, Fatima Siddiqui, Muhammad Khuzzaim Khan, Sindhura Tadisetty, Prasanna Kumar Gangishetti
Brain & Neurorehabilitation.2023;[Epub] CrossRef - Neurodegeneration with Brain Iron Accumulation and a Brief Report of the Disease in Iran
Reza Hajati, Maziar Emamikhah, Fardad Danaee Fard, Mohammad Rohani, Afagh Alavi
Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques.2022; 49(3): 338. CrossRef - A Practical Approach to Early-Onset Parkinsonism
Giulietta M. Riboldi, Emanuele Frattini, Edoardo Monfrini, Steven J. Frucht, Alessio Di Fonzo
Journal of Parkinson's Disease.2022; 12(1): 1. CrossRef - Parkinsonism and tremor syndromes
Steven Bellows, Joseph Jankovic
Journal of the Neurological Sciences.2022; 433: 120018. CrossRef - Aceruloplasminemia: MRI and Biochemical Profile Clue to Early Diagnosis in an Adolescent
Swapnil Sheth, Seema Sud, Tarvinder B. S. Buxi, Salil Bhargava, Ratna Dua Puri, Sapna Sandal, C.S. Agrawal
Journal of Pediatric Neurology.2022; 20(02): 133. CrossRef - Lifetime risk of autosomal recessive neurodegeneration with brain iron accumulation (NBIA) disorders calculated from genetic databases
Hana Kolarova, Jing Tan, Tim M. Strom, Thomas Meitinger, Matias Wagner, Thomas Klopstock
eBioMedicine.2022; 77: 103869. CrossRef - Two cases with mitochondrial membrane protein-associated neurodegeneration: genetic features and long-term clinical follow-up
Sevcan Mercan, Sibel Aylin Ugur Iseri, Remzi Yigiter, Nihan Hande Akcakaya, Esen Saka, Zuhal Yapici
Neurocase.2022; 28(1): 37. CrossRef - CHARACTERISTIC UPPER LIMB DYSTONIA PATTERN AS A CORRELATE TO THE 'EYE OF TIGER' MRI SIGN.
Akash Chheda, Dnyaneshwar V Jadhav, Sangeeta Ravat
GLOBAL JOURNAL FOR RESEARCH ANALYSIS.2022; : 28. CrossRef - A case of senile-onset progressive hemiballism and cognitive decline with diffuse brain iron accumulations
I-Ting Lin, Ni-Chung Lee, Sung-Pin Fan, Chang-Jin Huang, PoWei Cheng, Jyh-Horng Chen, Chin-Hsien Lin
Parkinsonism & Related Disorders.2022; 98: 114. CrossRef - A Case of MPAN with “Eye of the Tiger Sign,” Mimicking PKAN
Masoumeh Dehghan Manshadi, Mohammd Rohani, Ali Rezaei, Omid Aryani
Movement Disorders Clinical Practice.2022; 9(5): 693. CrossRef - Spinal cord stimulation for freezing of gait in Parkinson's disease and progressive supranuclear palsy: a case series
Vladislav V. Kovalev, Ekaterina V. Bril, Maksim S. Semenov, Yury A. Seliverstov, Levan T. Lepsveridze
Almanac of Clinical Medicine.2022; 50(5): 315. CrossRef - Early Neuroimaging Markers in β Propeller Protein-Associated Neurodegeneration
L. Chiapparini, G. Zorzi
American Journal of Neuroradiology.2022; 43(12): 1815. CrossRef - Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury‐induced ferroptosis
Tongyu Rui, Haochen Wang, Qianqian Li, Ying Cheng, Yuan Gao, Xuexian Fang, Xuying Ma, Guang Chen, Cheng Gao, Zhiya Gu, Shunchen Song, Jian Zhang, Chunling Wang, Zufeng Wang, Tao Wang, Mingyang Zhang, Junxia Min, Xiping Chen, Luyang Tao, Fudi Wang, Chengli
Journal of Pineal Research.2021;[Epub] CrossRef - Correlation of dystonia severity and iron accumulation in Rett syndrome
Tz-Yun Jan, Lee-Chin Wong, Ming-Tao Yang, Chien-Feng Judith Huang, Chia-Jui Hsu, Steven Shinn-Forng Peng, Wen-Yih Isaac Tseng, Wang-Tso Lee
Scientific Reports.2021;[Epub] CrossRef - The Case of a Patient with Pantothenate Kinase-Associated Neurodegeneration Presenting with a Prolonged History of Stuttering Speech and a Misdiagnosis of Parkinson’s Disease
Prashant A Natteru, Juebin Huang
Journal of Movement Disorders.2021; 14(1): 86. CrossRef - Retrospective analysis of 17 patients with mitochondrial membrane protein-associated neurodegeneration diagnosed in Russia
Peter Sparber, Tatiana Krylova, Svetlana Repina, Nina Demina, Galina Rudenskaya, Inna Sharkova, Artem Sharkov, Vitaly Kadyshev, Ilya Kanivets, Sergey Korostelev, Ekaterina Pomerantseva, Vladimir Kaimonov, Svetlana Mikhailova, Ekaterina Zakharova, Mikhail
Parkinsonism & Related Disorders.2021; 84: 98. CrossRef - Arching deep brain stimulation in dystonia types
Han-Joon Kim, Beomseok Jeon
Journal of Neural Transmission.2021; 128(4): 539. CrossRef -
WDR45 Gene and Its Role in Pediatric Epilepsies
Federica Filosco, Sebastiano Billone, Ausilia Collotta, Tiziana Timpanaro, Monica Tosto, Raffaele Falsaperla, Silvia Marino, Antonio Zanghì, Andrea D. Praticò
Journal of Pediatric Neurology.2021;[Epub] CrossRef - Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders
Vassilena Iankova, Ivan Karin, Thomas Klopstock, Susanne A. Schneider
Frontiers in Neurology.2021;[Epub] CrossRef - Neurodegeneration with brain iron accumulation: Characterization of clinical, radiological, and genetic features of pediatric patients from Southern India
Naveen Kumar Bhardwaj, Vykuntaraju K. Gowda, Jitendra Saini, Ashwin Vivek Sardesai, Rashmi Santhoshkumar, Anita Mahadevan
Brain and Development.2021; 43(10): 1013. CrossRef - Autophagic defects observed in fibroblasts from a patient with β‐propeller protein‐associated neurodegeneration
Jae‐Hyeok Lee, Sang Ook Nam, Eun Kyoung Kim, Jin‐Hong Shin, Seung Hwan Oh, Dongryeol Ryu, Hye Eun Lee, Ji Young Mun
American Journal of Medical Genetics Part A.2021; 185(12): 3866. CrossRef - Consensus clinical management guideline for beta‐propeller protein‐associated neurodegeneration
Jenny L Wilson, Allison Gregory, Manju A Kurian, Ittai Bushlin, Fanny Mochel, Lisa Emrick, Laura Adang, Penelope Hogarth, Susan J Hayflick
Developmental Medicine & Child Neurology.2021; 63(12): 1402. CrossRef - Findings of Magnetic Resonance and Dopamine Transporter Imaging in Beta-Propeller Protein-Associated Neurodegeneration
Seong Ho Jeong
Journal of the Korean Neurological Association.2021; 39(3): 239. CrossRef - An Eye on Movement Disorders
Duncan Wilson, Mark Hallett, Tim Anderson
Movement Disorders Clinical Practice.2021; 8(8): 1168. CrossRef - Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation
Robert V.V. Spaull, Audrey K.S. Soo, Penelope Hogarth, Susan J. Hayflick, Manju A. Kurian
Tremor and Other Hyperkinetic Movements.2021;[Epub] CrossRef - Novel C19orf12 loss-of-function variant leading to neurodegeneration with brain iron accumulation
Antonia Lefter, Iulia Mitrea, Dan Mitrea, Vasilica Plaiasu, Aida Bertoli-Avella, Christian Beetz, Liviu Cozma, Delia Tulbă, Cristina Elena Mitu, Bogdan Ovidiu Popescu
Neurocase.2021; 27(6): 481. CrossRef - Early-onset presentation of a new subtype of β-Propeller protein-associated neurodegeneration (BPAN) caused by a de novo WDR45 deletion in a 6 year-old female patient
Stephanie Christoforou, Kyproula Christodoulou, Violetta Anastasiadou, Paola Nicolaides
European Journal of Medical Genetics.2020; 63(3): 103765. CrossRef - A rare PANK2 deletion in the first north African patient affected with pantothenate kinase associated neurodegeneration
Stephanie Efthymiou, Yamna Kriouile, Vincenzo Salpietro, Rhouda Hajar, Zouiri Ghizlane, Kshitij Mankad, Mohamed El Khorassani, Mhammed Aguennouz, Henry Houlden, Sarah Wiethoff
Journal of the Neurological Sciences.2020; 410: 116639. CrossRef - Chemical Chaperones as Novel Drugs for Parkinson’s Disease
Jordi Pujols, Samuel Peña-Díaz, Irantzu Pallarès, Salvador Ventura
Trends in Molecular Medicine.2020; 26(4): 408. CrossRef - A case of novel WDR45 mutation with beta-propeller protein-associated neurodegeneration (BPAN) presenting asymmetrical extrapyramidal signs
Ryota Sato, Michiaki Koga, Kazuhiro Iwama, Tsuyoshi Mizuguchi, Naomichi Matsumoto, Takashi Kanda
Rinsho Shinkeigaku.2020; 60(5): 317. CrossRef - Atypical pantothenate kinase-associated neurodegeneration with PANK2 mutations : clinical description and a review of the literature
Si Pan, Chenkai Zhu
Neurocase.2020; 26(3): 175. CrossRef - The roles of iron and HFE genotype in neurological diseases
Yunsung Kim, James R. Connor
Molecular Aspects of Medicine.2020; 75: 100867. CrossRef - L'acéruléoplasminémie héréditaire, une pathologie à ne pas méconnaître
H. Lobbes, Q. Reynaud, S. Mainbourg, J-C. Lega, I. Durieu, S. Durupt
La Revue de Médecine Interne.2020; 41(11): 769. CrossRef - Atypical brain MRI in neurological Wilson disease
Jong Hyeon Ahn, Joomee Song, Inyoung Choi, Ji Sun Kim, Jin Whan Cho, Jinyoung Youn
Parkinsonism & Related Disorders.2020; 78: 70. CrossRef - Clinical features and blood iron metabolism markers in children with beta-propeller protein associated neurodegeneration
Anezka Belohlavkova, Katalin Sterbova, Cornelia Betzler, Stuve Burkhard, Axel Panzer, Markus Wolf, Petra Lassuthova, Marketa Vlckova, Martin Kyncl, Barbora Benova, Alena Jahodova, Martin Kudr, Maria Goerg, Petr Dusek, Pavel Seeman, Gerhard Kluger, Pavel K
European Journal of Paediatric Neurology.2020;[Epub] CrossRef - Loss of tyrosine hydroxylase, motor deficits and elevated iron in a mouse model of phospholipase A2G6-associated neurodegeneration (PLAN)
Michael Minkley, Patrick MacLeod, Christopher K. Anderson, Raad Nashmi, Patrick B. Walter
Brain Research.2020; 1748: 147066. CrossRef - Nanotechnology-based Targeting of Neurodegenerative Disorders: A Promising Tool for Efficient Delivery of Neuromedicines
Kuldeep Rajpoot
Current Drug Targets.2020; 21(8): 819. CrossRef - Brain MRI Pattern Recognition in Neurodegeneration With Brain Iron Accumulation
Jae-Hyeok Lee, Ji Young Yun, Allison Gregory, Penelope Hogarth, Susan J. Hayflick
Frontiers in Neurology.2020;[Epub] CrossRef - Molecular Defects in Friedreich’s Ataxia: Convergence of Oxidative Stress and Cytoskeletal Abnormalities
Frances M. Smith, Daniel J. Kosman
Frontiers in Molecular Biosciences.2020;[Epub] CrossRef - Neuroimaging of Basal Ganglia in Neurometabolic Diseases in Children
Justyna Paprocka, Magdalena Machnikowska-Sokołowska, Katarzyna Gruszczyńska, Ewa Emich-Widera
Brain Sciences.2020; 10(11): 849. CrossRef - Eye of tiger sign in patient with extrapyramidal syndrome - unique case report
Martin Daniš, Juraj Cisár, Georgi Krastev
Neurologie pro praxi.2020; 21(4): 319. CrossRef - Early onset developmental delay and epilepsy in pediatric patients with WDR45 variants
Hongbo Chen, Yanyan Qian, Sha Yu, Deyong Xiao, Xiao Guo, Qing Wang, Lili Hao, Kai Yan, Yulan Lu, Xinran Dong, Wenhao Zhou, Bingbing Wu, Shuizhen Zhou, Huijun Wang
European Journal of Medical Genetics.2019; 62(2): 149. CrossRef - A new NBIA patient from Turkey with homozygous C19ORF12 mutation
Çiğdem Seher Kasapkara, Leyla Tümer, Allison Gregory, Fatih Ezgü, Aslı İnci, Betül Emine Derinkuyu, Rachel Fox, Caleb Rogers, Susan Hayflick
Acta Neurologica Belgica.2019; 119(4): 623. CrossRef - Substantia Nigra Swelling and Dentate Nucleus T2 Hyperintensity May Be Early Magnetic Resonance Imaging Signs of β‐Propeller Protein‐Associated Neurodegeneration
Camilla Russo, Anna Ardissone, Elena Freri, Serena Gasperini, Marco Moscatelli, Giovanna Zorzi, Celeste Panteghini, Barbara Castellotti, Barbara Garavaglia, Nardo Nardocci, Luisa Chiapparini
Movement Disorders Clinical Practice.2019; 6(1): 51. CrossRef - A Golgi-targeting fluorescent probe for labile Fe(ii) to reveal an abnormal cellular iron distribution induced by dysfunction of VPS35
Tasuku Hirayama, Masatoshi Inden, Hitomi Tsuboi, Masato Niwa, Yasuhiro Uchida, Yuki Naka, Isao Hozumi, Hideko Nagasawa
Chemical Science.2019; 10(5): 1514. CrossRef - Iron in Neurodegeneration – Cause or Consequence?
Alain Ndayisaba, Christine Kaindlstorfer, Gregor K. Wenning
Frontiers in Neuroscience.2019;[Epub] CrossRef - Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis
Giacomo Marchi, Fabiana Busti, Acaynne Lira Zidanes, Annalisa Castagna, Domenico Girelli
Frontiers in Neuroscience.2019;[Epub] CrossRef - FAHN/SPG35: a narrow phenotypic spectrum across disease classifications
Tim W Rattay, Tobias Lindig, Jonathan Baets, Katrien Smets, Tine Deconinck, Anne S Söhn, Konstanze Hörtnagel, Kathrin N Eckstein, Sarah Wiethoff, Jennifer Reichbauer, Marion Döbler-Neumann, Ingeborg Krägeloh-Mann, Michaela Auer-Grumbach, Barbara Plecko, A
Brain.2019; 142(6): 1561. CrossRef - Juvenile parkinsonism: Differential diagnosis, genetics, and treatment
Nicki Niemann, Joseph Jankovic
Parkinsonism & Related Disorders.2019; 67: 74. CrossRef - Woodhouse–Sakati Syndrome: First report of a Portuguese case
Pedro Louro, João Durães, Diana Oliveira, Sandra Paiva, Lina Ramos, Maria Carmo Macário
American Journal of Medical Genetics Part A.2019; 179(11): 2237. CrossRef - Patterns of neurological manifestations in Woodhouse-Sakati Syndrome
Saeed Bohlega, Ali H. Abusrair, Fahad S. Al-Ajlan, Norah Alharbi, Abdulaziz Al-Semari, Balsam Bohlega, Dalya Abualsaud, Fowzan Alkuraya
Parkinsonism & Related Disorders.2019; 69: 99. CrossRef - Clinical case of a rare neurodegenerative disease with iron accumulation in the brain, type 4, in a 15-year-old child
I. F. Fedoseeva, T. V. Poponnikova, G. Yu. Galieva, S. V. Moschnegootz
Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics).2019; 64(5): 109. CrossRef - Levodopa-induced dyskinesias in mitochondrial membrane protein–associated neurodegeneration
Daniel Savitt, Joseph Jankovic
Neurology Clinical Practice.2019;[Epub] CrossRef - On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation
C. Tello, A. Darling, V. Lupo, B. Pérez‐Dueñas, C. Espinós
Clinical Genetics.2018; 93(4): 731. CrossRef - Les mouvements anormaux : mise au point
M. Béreau, C. Tranchant
La Revue de Médecine Interne.2018; 39(8): 641. CrossRef - Ferrosenescence: The iron age of neurodegeneration?
Adonis Sfera, Kelsey Bullock, Amy Price, Luzmin Inderias, Carolina Osorio
Mechanisms of Ageing and Development.2018; 174: 63. CrossRef - Chemodosimeters and chemoreactands for sensing ferric ions
Karl J. Wallace, Ashley D. G. Johnson, W. Scott Jones, Erendra Manandhar
Supramolecular Chemistry.2018; 30(5-6): 353. CrossRef - Beta‐propeller protein‐associated neurodegeneration: a case report and review of the literature
Kjersti Eline Stige, Ivar Otto Gjerde, Gunnar Houge, Per Morten Knappskog, Charalampos Tzoulis
Clinical Case Reports.2018; 6(2): 353. CrossRef - Looking Deep into the Eye-of-the-Tiger in Pantothenate Kinase–Associated Neurodegeneration
J.-H. Lee, A. Gregory, P. Hogarth, C. Rogers, S.J. Hayflick
American Journal of Neuroradiology.2018; 39(3): 583. CrossRef - Novel PLA2G6 mutations and clinical heterogeneity in Chinese cases with phospholipase A2-associated neurodegeneration
Yi-Jun Chen, Yu-Chao Chen, Hai-Lin Dong, Li-Xi Li, Wang Ni, Hong-Fu Li, Zhi-Ying Wu
Parkinsonism & Related Disorders.2018;[Epub] CrossRef - Expanding the phenotype of SLC25A42‐associated mitochondrial encephalomyopathy
M. Almannai, A. Alasmari, A. Alqasmi, E. Faqeih, F. Al Mutairi, M. Alotaibi, M.M. Samman, W. Eyaid, Y.I. Aljadhai, H.E. Shamseldin, W. Craigen, F.S. Alkuraya
Clinical Genetics.2018; 93(5): 1097. CrossRef - Basal ganglia calcification and novel compound heterozygous mutations in the PANK2 gene in a Chinese boy with classic Pantothenate kinase-associated neurodegeneration
Xulai Shi, Feixia Zheng, Xiuyun Ye, Xiucui Li, Qianlei Zhao, Zhongdong Lin, Ying Hu, Jiwen Wang
Medicine.2018; 97(15): e0316. CrossRef - Botulinum toxin injection to improve functional independence and to alleviate parenting stress in a child with advanced pantothenate kinase-associated neurodegeneration
Cho-I Lin, Kuan-Lin Chen, Ta-Shen Kuan, Sheng-Han Lin, Wei-Pin Lin, Yu-Ching Lin
Medicine.2018; 97(20): e10709. CrossRef - Expanding the Spectrum of Dopa-Responsive Dystonia (DRD) and Proposal for New Definition: DRD, DRD-plus, and DRD Look-alike
Woong-Woo Lee, Beomseok Jeon, Ryul Kim
Journal of Korean Medical Science.2018;[Epub] CrossRef - Oxidative stress and neurodegeneration: the involvement of iron
Alessia Carocci, Alessia Catalano, Maria Stefania Sinicropi, Giuseppe Genchi
BioMetals.2018; 31(5): 715. CrossRef - Non-cell-autonomous actions of α-synuclein: Implications in glial synucleinopathies
Somin Lim, Han-Joon Kim, Dong-Kyu Kim, Seung-Jae Lee
Progress in Neurobiology.2018; 169: 158. CrossRef - Longitudinal study of multiple sclerosis lesions using ultra-high field (7T) multiparametric MR imaging
Sanjeev Chawla, Ilya Kister, Tim Sinnecker, Jens Wuerfel, Jean-Christophe Brisset, Friedemann Paul, Yulin Ge, Quan Jiang
PLOS ONE.2018; 13(9): e0202918. CrossRef - Brain MR Imaging Findings in Woodhouse-Sakati Syndrome
A.H. Abusrair, S. Bohlega, A. Al-Semari, F.S. Al-Ajlan, K. Al-Ahmadi, B. Mohamed, A. AlDakheel
American Journal of Neuroradiology.2018; 39(12): 2256. CrossRef - The Involvement of Iron in Traumatic Brain Injury and Neurodegenerative Disease
Maria Daglas, Paul A. Adlard
Frontiers in Neuroscience.2018;[Epub] CrossRef - Imaging of dentate nucleus pathologies; a pictorial essay
Kajari Bhattacharya, Hima Pendharkar, Arun K Gupta
Indian Journal of Radiology and Imaging.2018; 28(02): 152. CrossRef - Identification of Novel Compound Mutations in PLA2G6-Associated Neurodegeneration Patient with Characteristic MRI Imaging
Sen Guo, Liu Yang, Huijie Liu, Wei Chen, Jinchen Li, Ping Yu, Zhong Sheng Sun, Xiang Chen, Jie Du, Tao Cai
Molecular Neurobiology.2017; 54(6): 4636. CrossRef - Serum Ferritin Levels Are Lower in Children With Tic Disorders Compared with Children Without Tics: A Cross-Sectional Study
Matan Avrahami, Ran Barzilay, Miki HarGil, Abraham Weizman, Nathan Watemberg
Journal of Child and Adolescent Psychopharmacology.2017; 27(2): 192. CrossRef - Autres syndromes parkinsoniens
Christine Tranchant
La Presse Médicale.2017; 46(2): 210. CrossRef - Ferrous Iron Up-regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN)
Rosaria Ingrassia, Maurizio Memo, Barbara Garavaglia
Frontiers in Genetics.2017;[Epub] CrossRef - Radiological Findings of Two Sisters with Aceruloplasminemia Presenting with Chorea
H. K. Kim, C. S. Ki, Y. J. Kim, M. S. Lee
Clinical Neuroradiology.2017; 27(3): 385. CrossRef - Mitochondrial membrane protein-associated neurodegeneration
Angela Deutschländer, Takuya Konno, Owen A. Ross
Parkinsonism & Related Disorders.2017; 39: 1. CrossRef - Intragenic deletion of the WDR45 gene in a male with encephalopathy, severe psychomotor disability, and epilepsy
Sylvia Redon, Caroline Benech, Sacha Schutz, Aurore Despres, Paul Gueguen, Pauline Le Berre, Cédric Le Marechal, Sylviane Peudenier, Philippe Meriot, Philippe Parent, Claude Ferec
American Journal of Medical Genetics Part A.2017; 173(5): 1444. CrossRef - Clinical and Imaging Presentation of a Patient with Beta-Propeller Protein-Associated Neurodegeneration, a Rare and Sporadic form of Neurodegeneration with Brain Iron Accumulation (NBIA)
Elke Hattingen, Nikolaus Handke, Kirsten Cremer, Sabine Hoffjan, Guido Matthias Kukuk
Clinical Neuroradiology.2017; 27(4): 481. CrossRef - Dentate Update: Imaging Features of Entities That Affect the Dentate Nucleus
K.M. Bond, W. Brinjikji, L.J. Eckel, D.F. Kallmes, R.J. McDonald, C.M. Carr
American Journal of Neuroradiology.2017; 38(8): 1467. CrossRef - Available treatment options for dystonia
Isabel Alfradique-Dunham, Joseph Jankovic
Expert Opinion on Orphan Drugs.2017; 5(9): 707. CrossRef - Clinical rating scale for pantothenate kinase‐associated neurodegeneration: A pilot study
Alejandra Darling, Cristina Tello, María Josep Martí, Cristina Garrido, Sergio Aguilera‐Albesa, Miguel Tomás Vila, Itziar Gastón, Marcos Madruga, Luis González Gutiérrez, Julio Ramos Lizana, Montserrat Pujol, Tania Gavilán Iglesias, Kylee Tustin, Jean Pie
Movement Disorders.2017; 32(11): 1620. CrossRef - iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease
Charles Arber, Plamena R. Angelova, Sarah Wiethoff, Yugo Tsuchiya, Francesca Mazzacuva, Elisavet Preza, Kailash P. Bhatia, Kevin Mills, Ivan Gout, Andrey Y. Abramov, John Hardy, James A. Duce, Henry Houlden, Selina Wray, Fanis Missirlis
PLOS ONE.2017; 12(9): e0184104. CrossRef - Presynaptic Dopaminergic Degeneration in a Patient with Beta-Propeller Protein-Associated Neurodegeneration Documented by Dopamine Transporter Positron Emission Tomography Images: A Case Report
Min Ki Kim, Nan Young Kim, Sangkyoon Hong, Hyeo-Il Ma, Yun Joong Kim
Journal of Movement Disorders.2017; 10(3): 161. CrossRef - Iron in neurodegenerative disorders: being in the wrong place at the wrong time?
Sotirios Apostolakis, Anna-Maria Kypraiou
Reviews in the Neurosciences.2017; 28(8): 893. CrossRef - Lessons from a pair of siblings with BPAN
Yuri A Zarate, Julie R Jones, Melanie A Jones, Francisca Millan, Jane Juusola, Annette Vertino-Bell, G Bradley Schaefer, Michael C Kruer
European Journal of Human Genetics.2016; 24(7): 1080. CrossRef - “Eye of tiger sign” mimic in an adolescent boy with mitochondrial membrane protein associated neurodegeneration (MPAN)
Sangeetha Yoganathan, Sniya Valsa Sudhakar, Maya Thomas, Atanu Kumar Dutta, Sumita Danda
Brain and Development.2016; 38(5): 516. CrossRef - Gene co-expression networks shed light into diseases of brain iron accumulation
Conceição Bettencourt, Paola Forabosco, Sarah Wiethoff, Moones Heidari, Daniel M. Johnstone, Juan A. Botía, Joanna F. Collingwood, John Hardy, Elizabeth A. Milward, Mina Ryten, Henry Houlden
Neurobiology of Disease.2016; 87: 59. CrossRef - Brain iron accumulation affects myelin-related molecular systems implicated in a rare neurogenetic disease family with neuropsychiatric features
M Heidari, D M Johnstone, B Bassett, R M Graham, A C G Chua, M J House, J F Collingwood, C Bettencourt, H Houlden, M Ryten, J K Olynyk, D Trinder, E A Milward
Molecular Psychiatry.2016; 21(11): 1599. CrossRef - WDR45 mutations in Rett (-like) syndrome and developmental delay: Case report and an appraisal of the literature
Sabine Hoffjan, Aysegül Ibisler, Anne Tschentscher, Gabriele Dekomien, Carla Bidinost, Alberto L. Rosa
Molecular and Cellular Probes.2016;[Epub] CrossRef - Missions of <italic>Journal of Movement Disorders</italic>
Yun Joong Kim
Journal of Movement Disorders.2016; 9(1): 1. CrossRef - Clinical Heterogeneity of Atypical Pantothenate Kinase-Associated Neurodegeneration in Koreans
Jae-Hyeok Lee, Jongkyu Park, Ho-Sung Ryu, Hyeyoung Park, Young Eun Kim, Jin Yong Hong, Sang Ook Nam, Young-Hee Sung, Seung-Hwan Lee, Jee-Young Lee, Myung Jun Lee, Tae-Hyoung Kim, Chul Hyoung Lyoo, Sun Ju Chung, Seong Beom Koh, Phil Hyu Lee, Jin Whan Cho,
Journal of Movement Disorders.2016; 9(1): 20. CrossRef - «Et høist mærkeligt Sygdomstilfælde hos flere Sødskende» – en norsk førstegangsbeskrivelse fra 1830?
Magne Nylenna, Noralv Breivik, Arvid Heiberg, Øivind Larsen
Tidsskrift for Den norske legeforening.2016; 136(5): 437. CrossRef - Woodhouse-Sakati Syndrome: Report of the First Tunisian Family with the C2orf37 Gene Mutation
Olfa Hdiji, Emna Turki, Nouha Bouzidi, Imen Bouchhima, Mariem Damak, Saeed Bohlega, Chokri Mhiri
Journal of Movement Disorders.2016; 9(2): 120. CrossRef - Encefalopatie metaboliche e tossiche non farmacologiche
P. Codron, C. Verny
EMC - Neurologia.2016; 16(3): 1. CrossRef - Iron and Non-Iron-Related Characteristics of Multiple Sclerosis and Neuromyelitis Optica Lesions at 7T MRI
S. Chawla, I. Kister, J. Wuerfel, J.- C. Brisset, S. Liu, T. Sinnecker, P. Dusek, E. M. Haacke, F. Paul, Y. Ge
American Journal of Neuroradiology.2016; 37(7): 1223. CrossRef - Freezing of gait in Parkinson's disease: from pathophysiology to emerging therapies
Alberto Cucca, Milton C Biagioni, Jori E Fleisher, Shashank Agarwal, Andre Son, Pawan Kumar, Miroslaw Brys, Alessandro Di Rocco
Neurodegenerative Disease Management.2016; 6(5): 431. CrossRef - Neurodegeneration with brain iron accumulation
João Carlos Papaterra Limongi
Arquivos de Neuro-Psiquiatria.2016; 74(7): 517. CrossRef - Neurodegeneration with brain iron accumulation: A case report
Daniel Nassif, João Santos Pereira, Mariana Spitz, Cláudia Capitão, Alessandra Faria
Dementia & Neuropsychologia.2016; 10(2): 160. CrossRef - What’s in a name? Problems, facts and controversies regarding neurological eponyms
Hélio A. G. Teive, Plínio M. G. Lima, Francisco M. B. Germiniani, Renato P. Munhoz
Arquivos de Neuro-Psiquiatria.2016; 74(5): 423. CrossRef - A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging
Rubens Paulo Araújo Salomão, José Luiz Pedroso, Maria Thereza Drumond Gama, Lívia Almeida Dutra, Ricardo Horta Maciel, Clécio Godeiro-Junior, Hsin Fen Chien, Hélio A. G. Teive, Francisco Cardoso, Orlando G. P. Barsottini
Arquivos de Neuro-Psiquiatria.2016; 74(7): 587. CrossRef - Novel homozygous PANK2 mutation identified in a consanguineous Chinese pedigree with pantothenate kinase-associated neurodegeneration
Yan-Fang Li, Hong-Fu Li, Yan-Bin Zhang, Ji-Min Wu
Biomedical Reports.2016; 5(2): 217. CrossRef - NBIA - neurodegeneration with brain iron accumulation
Lenka Hvizdošová, Michaela Kaiserová, Petr Kaňovský, Marek Baláž
Neurologie pro praxi.2016; 17(5): 328. CrossRef - Orally Bioavailable Metal Chelators and Radical Scavengers: Multifunctional Antioxidants for the Coadjutant Treatment of Neurodegenerative Diseases
Hiroyoshi Kawada, Peter F. Kador
Journal of Medicinal Chemistry.2015; 58(22): 8796. CrossRef - Voxel-based analysis in neuroferritinopathy expands the phenotype and determines radiological correlates of disease severity
M. J. Keogh, B. S. Aribisala, J. He, E. Tulip, D. Butteriss, C. Morris, G. Gorman, R. Horvath, P. F. Chinnery, Andrew M. Blamire
Journal of Neurology.2015; 262(10): 2232. CrossRef - A field guide to current advances in paediatric movement disorders
Laura Silveira-Moriyama, Jean-Pierre Lin
Current Opinion in Neurology.2015; 28(4): 437. CrossRef - En mann i 50-årene med høyt ferritinnivå og økende kognitiv svikt
Marte-Helene Bjørk, Ivar Otto Gjerde, Charalampos Tzoulis, Rune Johan Ulvik, Laurence Albert Bindoff
Tidsskrift for Den norske legeforening.2015; 135(15): 1369. CrossRef - Iron, Aging, and Neurodegeneration
Dafina Angelova, David Brown
Metals.2015; 5(4): 2070. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

