Ultrastructures of α-Synuclein Filaments in Synucleinopathy Brains and Experimental Models
Article information

Abstract
Intracellular α-synuclein (α-syn) inclusions are a neuropathological hallmark of Lewy body disease (LBD) and multiple system atrophy (MSA), both of which are termed synucleinopathies. LBD is defined by Lewy bodies and Lewy neurites in neurons, while MSA displays glial cytoplasmic inclusions in oligodendrocytes. Pathological α-syn adopts an ordered filamentous structure with a 5–10 nm filament diameter, and this conformational change has been suggested to be involved in the disease onset and progression. Synucleinopathies also exhibit characteristic ultrastructural and biochemical properties of α-syn filaments, and α-syn strains with distinct conformations have been identified. Numerous experimental studies have supported the idea that pathological α-syn self-amplifies and spreads throughout the brain, during which processes the conformation of α-syn filaments may drive the disease specificity. In this review, we summarize the ultrastructural features and heterogeneity of α-syn filaments in the brains of patients with synucleinopathy and in experimental models of seeded α-syn aggregation.
INTRODUCTION
Synucleinopathies are a subset of neurodegenerative diseases characterized by inclusions in which α-synuclein (α-syn) is a major component. In Lewy body disease (LBD), including Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB), Lewy bodies (LBs) characterized by an eosinophilic core and a surrounding clear halo are observed in neurons, and these structures are commonly described as brainstem-type LBs (Figure 1A) [1-3]. Cortical-type LBs lacking a distinctive core and halo are also observed in LBD, as well as Lewy neurites (LNs) in axons and dendrites (Figure 1A) [1,4]. In 1997, the A53T mutation in the SNCA gene encoding α-syn was reported to cause familial PD in Italian and Greek families [5]. This finding led to the identification of α-syn as a major component of LBs [6,7]. In 1998, it was further established that α-syn composes glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA) (Figure 1A) [8]. MSA is divided into two types based on motor phenotype: parkinsonian type (MSA-P), with predominant extrapyramidal symptoms, and cerebellar type (MSA-C), characterized by cerebellar dysfunction [9]. Currently [10], missense mutations (A30P, A30G, E46K, H50Q, G51D, A53T, A53E, A53V, T72M, E83Q) and multiplication (duplication, triplication) of the SNCA gene have been reported to be linked to the onset of synucleinopathy (Figure 1B) [5,10-20]. The above discoveries paved the way for the detailed characterization of α-syn-related pathogenesis both in vitro and in vivo.
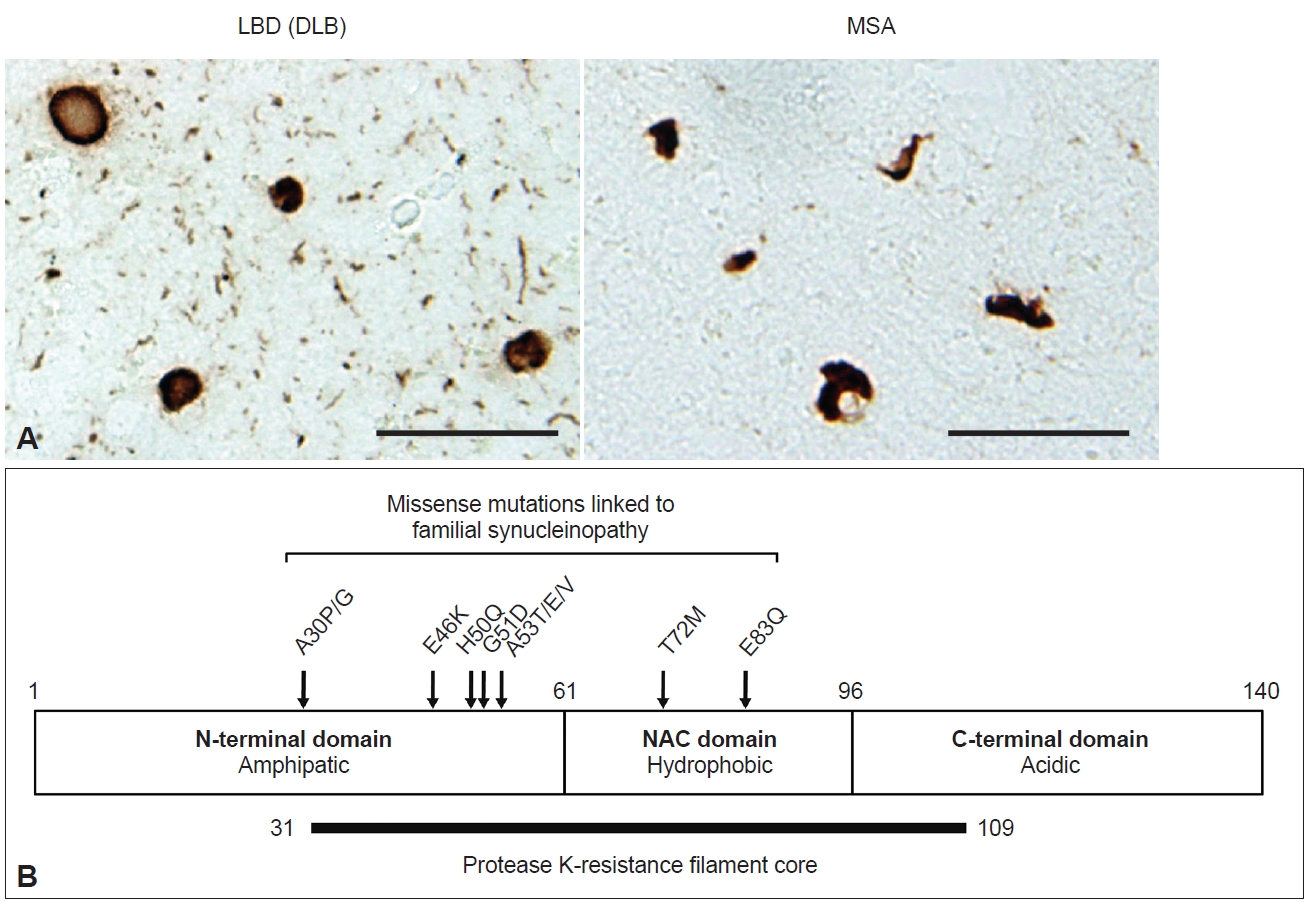
α-syn pathologies in LBD and MSA. A: Lewy bodies and Lewy neurites in a DLB case (left) and glial cytoplasmic inclusions in an MSA case (right) detected by pSer129 antibody. Scale bar, 50 μm. Data are original for this review. B: Schematic representation of human α-syn. The structure of α-syn consists of three domains: an N-terminal domain (1–60 residues), a non-β amyloid component (NAC, 61–95 residues), and a C-terminal domain (96–140 residues). The box above represents missense mutations associated with the onset of synucleinopathy. Protease K treatment of recombinant α-syn filaments indicates the existence of a filament core composed of 31–109 residues. LBD, Lewy body disease; DLB, dementia with Lewy bodies; MSA, multiple system atrophy.
α-syn is a small protein consisting of 140 amino acids, with an amphipathic N-terminal domain (1–60 residues), a non-β amyloid component (NAC) domain (61–95 residues) and an acidic C-terminal domain (96–140 residues) (Figure 1B). The N-terminal domain adopts an α-helical structure, and seven 11-amino-acid imperfect repeats containing the KTEGV motif are found in the N-terminal and NAC domains [21,22]. The NAC domain, originally identified as peptides X and Y in the sodium dodecyl sulfate (SDS)-insoluble fraction prepared from Alzheimer’s disease (AD) brains [23], is hydrophobic and responsible for the amyloidogenic property of α-syn [24]. The C-terminal region is negatively charged, and its truncation (121–140 residues) enhances the cytotoxicity and aggregation of α-syn [25-27]. α-syn is highly expressed in neurons, accounting for approximately 1% of total proteins expressed in the brain, and is localized to presynaptic terminals [28]. Although its physiological function is uncertain, studies of α-syn-knockout mice indicate that α-syn is involved in dopamine release [29], and roles in the regulation of neurotransmitter release and synaptic plasticity through chaperoning SNARE complex assembly and clustering synaptic vesicles have been proposed [30-32]. While α-syn also has a potential role in the regulation of lipid metabolism [33-35], mutations in related genes, such as GBA1 and VSP35, are the most plausible genetic risk factors for LBD onset [36,37]. Under physiological conditions, α-syn is categorized as a natively unfolded protein and is highly water soluble [38]. In contrast, pathological α-syn extracted from synucleinopathy brains is conformationally converted into β-sheet-rich amyloid-like filaments, which are insoluble in detergents such as sarkosyl, SDS and Triton X-100 and exhibit resistance to proteases [39-43]. These properties are akin to those of the infectious prion protein that causes prion disease [44]. Protease K (PK) treatment of recombinant α-syn filaments revealed that residues 31–109 form the filament core (Figure 1B) [42]. Most missense mutations are located in this region, suggesting that structural changes in normal α-syn cause rapid aggregation and result in early disease onset. Phosphorylation at S129 (pS129) is detected at > 90% frequency in synucleinopathy brains [45] and has been widely used as a marker to distinguish normal α-syn from pathological α-syn in both clinical and research fields. In addition to pS129, phosphorylation at other residues, such as Y39, T59, T64, T72, T81, Y125, Y133, and Y136, ubiquitination at K6, K12, K23, K60, and K80, N-terminal acetylation, O-GlcNAcylation, nitration and C-terminal truncation have also been reported in pathological α-syn [27,46-49].
Furthermore, α-syn filaments extracted from synucleinopathy brains and recombinant α-syn filaments have been demonstrated to work as “seeds” to trigger α-syn aggregation and pathology formation in in vitro and cellular systems and in animals, suggesting that the disease progression in LBD and MSA is based on prion-like amplification and spreading of pathological α-syn [50-52]. Disease-specific α-syn filaments recruit normal α-syn into a filament form in a templated manner and self-amplify (Figure 2). The amplified α-syn filaments then transmit from cell to cell and spread throughout the brain along neuronal networks (Figure 2). In these processes, disease specificity is thought to be maintained through inheritance of the conformation of the original seeds. Although the molecular mechanisms remain unclear, experimental studies have supported the idea that this prion-like hypothesis can explain the stereotypical expansion of α-syn pathology in LBD brains [53,54]. The observations of Lewy pathology in fetal dopamine neurons 10–24 years after transplantation into PD patients are also consistent with cell-to-cell transmission of pathological α-syn [55-57]. In recent years, cryo-electron microscopy (EM) structural analysis of patient-derived α-syn filaments has substantially advanced the understanding of the structural heterogeneity of the inclusions in synucleinopathies [48,58,59]. This review describes the characteristics of α-syn filaments accumulated in synucleinopathy brains and formed in experimental seeded aggregation models.
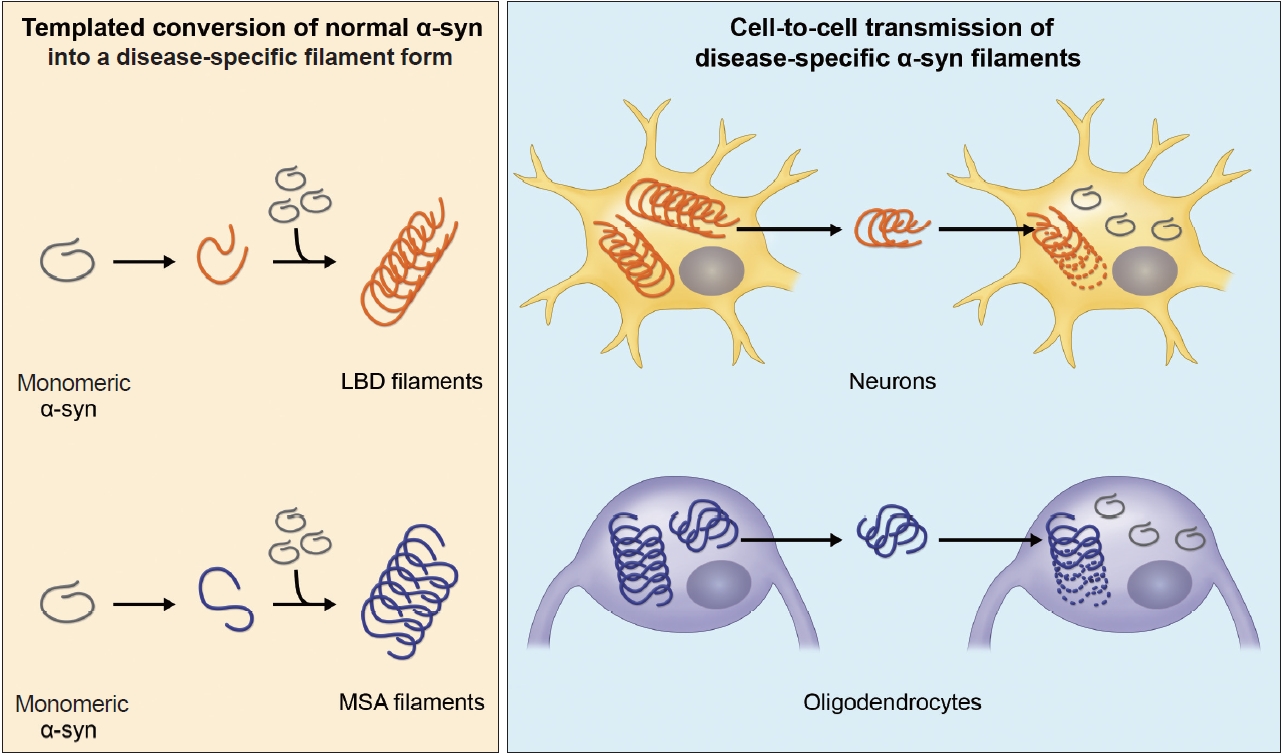
Formation and cell-to-cell transmission of disease-specific α-syn filaments in LBD and MSA.Under pathological conditions, monomeric α-syn is transformed to a misfolded form by factor(s) such as cellular environments and post-translational modifications, and the misfolded α-syn stacks regularly and forms α-syn filaments. The mode of misfolding differs depending on the disease, and the disease-specific α-syn filaments self-amplify. Furthermore, α-syn filaments accumulated in cells are released into the extracellular space and taken up into neighboring cells, leading to intracerebral spreading of α-syn pathology. Transmission of pathological α-syn between neurons has been experimentally demonstrated, whereas α-syn pathogenesis in oligodendrocytes remains unclear. LBD, Lewy body disease; MSA, multiple system atrophy.
α-SYN FILAMENTS IN SYNUCLEINOPATHY BRAINS
Duffy and Tennyson [60] first reported in 1965 that LBs observed in PD are composed of filamentous structures. EM of LBD brain sections revealed that abundant 7–20 nm straight filaments and their bundles fill LBs, and are distinct from neurofilaments having side arms [60,61]. Furthermore, brainstem-type LBs form a dense central core with radiating filaments, whereas the filaments in cortical-type LBs are less organized and more diffuse [4]. Besides the filaments, which later turned out to contain α-syn, the presence of lipids, membrane fragments, globular materials, and partial cytoplasmic organelles in LBs was also described [60,62]. Recently, stimulated emission depletion-based superresolution microscopy demonstrated the high occupancy of lipids and organelles in Lewy pathology [63]. More than 100 proteins, including proteins related to proteolytic systems, oxidative stress, cell signaling and protein folding, have been reported to colocalize with α-syn in LBs [64-66]. GCIs in MSA brain sections are constructed of filaments approximately 25 nm in diameter, which are not tightly associated with the cell membranes [67,68]. α-syn filaments accumulated in synucleinopathy brains can be extracted by fractionation using detergents. Abundant filaments labeled with α-syn antibodies are observed in detergent-insoluble fractions [7]. The ultrastructures of the extracted filaments resemble those observed in the brain sections. LBD-derived α-syn filaments (LBD-α-syn) are observed as linear structures 5–10 nm in diameter, whereas MSA-derived α-syn filaments (MSA-α-syn) have the same diameter but exhibit a twist with 80–100 nm periodicity (Figure 3A) [69-71]. The C-terminal α-syn antibody labels the entirety of both α-syn filaments, whereas the N-terminal α-syn antibody recognizes only one side of the filament ends, suggesting that α-syn filaments have a polar structure [69,70,72]. The ultrastructural differences between LBD-α-syn and MSA-α-syn are also demonstrated by protease digestion of the nonfilament core region. PK treatment shows disease-specific PK-resistant α-syn banding patterns, indicating that LBD-α-syn and MSA-α-syn adopt distinct filament core structures [73]. Furthermore, MSA-α-syn exhibits higher SDS solubility than LBD-α-syn [40,74].
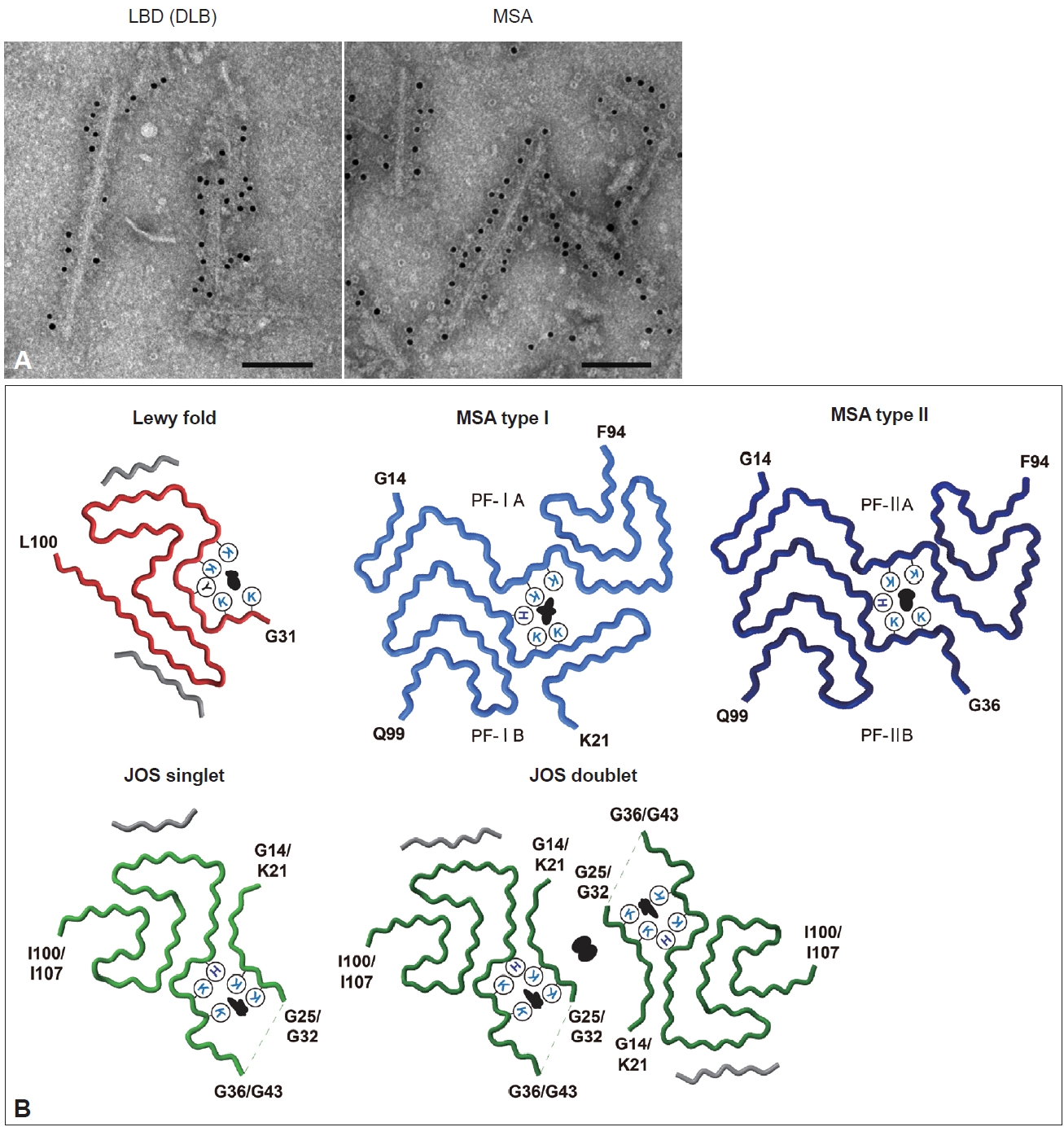
Structures of α-syn filaments extracted from synucleinopathy brains. A: Anti-C-terminal-α-syn-positive (syn131–140) filaments extracted from the brains of patients with DLB (left) and MSA (right) were immunolabeled with secondary antibody conjugated to 5 nm gold nanoparticles. Scale bar, 100 nm. Data are original for this review. B: The cryo-EM structures of α-syn filaments extracted from synucleinopathy brains. The Lewy fold (PDB ID: 8A9L) was identified from PD, PDD, and DLB cases. Two types of MSA α-syn filaments (PDB ID: 6XYO, 6XYP) were identified from MSA-P and MSA-C cases. One JOS case showed singlet and doublet α-syn filaments with the JOS fold (PDB ID: 8BQV, 8BQW). Black indicates additional density surrounded by lysine (K), histidine (H), and tyrosine (Y) residues or between protofilaments. Molecular graphics were created with UCSF ChimeraX[176]. LBD, Lewy body disease; DLB, dementia with Lewy bodies; MSA, multiple system atrophy; EM, electron microscopy; PDB, Protein Data Bank; PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; MSA-P, parkinsonian type; MSA-C, cerebellar type; JOS, juvenile-onset synucleinopathy.
In the last few years, structural polymorphisms of patient-derived α-syn filaments have been demonstrated at the atomic level by single-particle analysis using cryo-EM [48,58,59]. Two types of asymmetrical structures of α-syn filaments extracted from the putamen of five MSA cases, including MSA-P and MSA-C, were reported in 2020 (Figure 3B) [48]. These Type I and Type II filaments were each composed of two protofilaments with distinct filament cores. Protofilaments of Type I consist of G14-F94 (PF-IA) and K21-Q99 (PF-IB); the N-terminal region of PF-IA has an elongated one-layer L-shaped motif that is absent in PF-IB, and the C-terminal regions of both protofilaments show a compact three-layer L-shaped motif but with different packing. Type II filaments are made of protofilaments comprising G14-F94 (PF-IIA) and G36-Q99 (PF-IIB), which differ from those of Type I filaments. PF-IA and PF-IIA or PF-IB and PF-IIB show similar core structures, but PF-IIA contains a more extended cavity in the C-terminal region, and PF-IIB has a smaller N-terminal arm. Two types of substructures were identified at A78-Q99 in PF-IIB. In addition, a negatively charged additional density surrounded by K43, K45 and H50, which is distinguished from α-syn, is present between the two protofilaments of Type I and Type II. This suggests that some unidentified nonproteinaceous molecules are involved in the packing of the two protofilaments. The ratio of Type I to Type II in the putamen varied from case to case, and this variation was also observed in different brain regions of a single MSA case. Although the disease duration and the presence of neuronal cytoplasmic inclusions potentially alter the ratio of Type I to Type II, further investigation is needed to clarify what factors, such as brain regions, cell types, and other cellular environmental factors, influence this issue. In 2022, α-syn filaments extracted from the cingulate cortex or frontal cortex of six LBD cases, including PD, PDD, and DLB, were reported to show a common protofilament core structure termed the Lewy fold (Figure 3B) [58]. Most LBD-derived α-syn filaments are ultrastructurally straight, but the Lewy fold was determined from the twisted parts seen in approximately 25% of filaments. The Lewy fold consists of G31-L100, and nine β-strands form a three-layered core structure. Two islands with unknown amino acid sequences are packed in the Lewy fold. An additional density surrounded by K32, K34, Y39, K43 and K45, resembling in size those found in MSA, was also identified. Cryo-EM analysis of α-syn filaments derived from MSA and LBD brains showed that α-syn filaments with one or multiple common folds form disease-characteristic α-syn pathology, indicating that synucleinopathy can be defined in terms of α-syn filament structure. Since peripheral α-syn pathology is prominent in patients with LBD [75-78], it will also be important to determine the atomic structure of α-syn filaments derived from peripheral tissues. More recently, the juvenile-onset synucleinopathy (JOS) fold, which differs from the MSA folds and the Lewy fold, was identified in α-syn filaments extracted from one JOS case that developed DLB at 13 years of age (Figure 3B) [59,79]. Genetic analysis found a 7-amino-acid insertion (MAAAEKT) after T22, resulting from a 21-nucleotide duplication in one of the SNCA alleles. Immunoblot analysis of the detergent-insoluble fraction of this case detected bands of both wild-type (WT) α-syn (15 kDa) and insertion mutant α-syn comprising 147 amino acids (16 kDa). The JOS fold consists of G36-I100 in WT α-syn or G43-I107 in insertion mutant α-syn (the main core is not affected by the insertion) and two disconnected islands. Protofilaments and C2 symmetrically packed doublets were identified at a ratio of approximately 4:1. Partial similarity with the substructure common to Type I and Type II in MSA is found in the main core of the JOS fold. One island packed at the N-terminus of the main core appears to consist of G14-G25 in WT α-syn or K21-G32 in insertion mutant α-syn, and this island is probably essential for the JOS doublet structure. An additional density surrounded by K21/K28, K23/K30, K43/K50, K45/K52, and H50/H57 was present between this island and the main core. WT α-syn expressed in this JOS case did not adopt the Lewy fold, and WT and insertion mutant α-syn filaments showed a common fold. This may imply that insertion mutant α-syn first formed the JOS fold, then worked as a template, and recruited WT α-syn into the JOS fold form. Cryo-EM structures of patient-derived α-syn filaments provide evidence not only for the presence of α-syn strains in synucleinopathy, but also for templated amplification of disease-specific α-syn filaments in human brains.
α-SYN FILAMENTS IN IN VITRO MODELS
Experimental seeded α-syn aggregation has been demonstrated in vitro and in cultured cells, as well as in primary cultures and in animals. Recombinant α-syn purified from Escherichia coli forms amyloid-like filaments 5–10 nm in diameter after shaking at 37°C for several days. Mutant and truncated α-syn filaments exhibit different ultrastructural and biochemical properties from WT filaments. Most α-syn mutants have been reported to show a faster increase in thioflavin fluorescence intensity compared to WT, though A30P and G51D α-syn show a slower increase [80-85]. A30P α-syn filaments are more fragile than WT filaments, and seeded A30P filaments generated using WT monomers are also fragile [82]. C-terminally truncated α-syn forms shorter, more twisted filaments, and filament formation is relatively rapid compared to that of full-length α-syn [86-88]. These various types of recombinant α-syn filaments exhibit distinct protease-resistant α-syn banding patterns [82], supporting the idea that conformational differences give rise to different prion-like properties. In addition, recombinant α-syn filament strains can be formed from identical α-syn monomers, depending on the buffer composition, the presence of cofactors, and other conditions, such as shaking and temperature. The formation of ribbon-like or straight α-syn filaments is induced in the presence or absence of inorganic salts, and these strains exhibit different aggregation kinetics and interactions with the 26S proteasome [89,90]. Glycosaminoglycans such as heparin, metal ions and lipid vesicles also promote α-syn aggregation, leading to structural diversity [91-93]. The structures of recombinant WT, mutant, and truncated α-syn filaments have been determined by cryo-EM analysis [94-105]. Recombinant α-syn filaments have no structural identity at the atomic level with patient-derived α-syn filaments and show variable filament cores according to mutations and in vitro conditions. However, the substructures of patient-derived filaments and several types of recombinant filaments share similarities. In particular, V48-T92 containing the NAC domain tends to adopt a three-layer L-shaped structure, which may be due to a smaller nonfilament core region.
Additionally, amplification of α-syn contained in tissues and body fluids from patients with synucleinopathy has been utilized to develop the in vitro seed amplification assay (SAA), also known as PMCA or RT-QuIC [106,107]. Recombinant α-syn monomers and patient-derived samples are mixed and shaken at constant intervals during incubation. In general, different kinetics of thioflavin fluorescence intensities are seen in LBD and MSA [108,109], which may represent distinct bindings of thioflavin to the SAA product. Although SAA is undoubtedly valuable for the early diagnosis of synucleinopathy [110], the importance of the resulting α-syn filaments is currently questionable. SAA was performed using sarkosyl-insoluble fractions extracted from three MSA cases used for cryo-EM study [48], and the structures of the SAA products were identified [111]. Some types of the amplified filaments resembled PF-IIB, but MSA filaments were not reproduced in SAA. Other types of filaments showed structures resembling those of spontaneously formed recombinant α-syn filaments. Cerebrospinal fluid collected from PD patients at various disease stages was also used for SAA, and the structures of the SAA products were reported [112]. As in the study described earlier, the majority of amplified α-syn filaments were identical to spontaneous recombinant α-syn filaments, and several other filaments identified at low percentages did not show the Lewy fold. The structures of α-syn filaments formed in other SAA studies also differed from those of brain-derived filaments [113-116]. More recently, it has been demonstrated that α-syn in serum from patients with synucleinopathy can be amplified by SAA after immunoprecipitation [117]. The ultrastructures of SAA products differed from those of brain-derived filaments, such as straight or twisted protofilaments in MSA-derived products and twisted or bundled filaments in LBD-derived products. Notably, the structures identified from SAA products do not necessarily reflect the structures of α-syn in body fluids. Since the α-syn concentration in body fluids is extremely low, at the picomolar level [118], the molecular species and structural details of body fluid-derived α-syn have not yet been fully established. Structural interpretation of α-syn filaments obtained from SAA using body fluids requires further characterization of the original seeds.
α-SYN FILAMENTS IN CELLULAR MODELS
As in in vitro studies, seeded α-syn aggregation also occurs in cultured cells and rodent primary cultures, and intracellular accumulation of filamentous α-syn is observed in seeded cells [119-122]. A few days after introduction of recombinant human α-syn filaments into neuroblastoma SH-SY5Y cells expressing human WT α-syn, introduced by lipofection, pS129-positive α-syn inclusions resembling those observed in the patient’s brain were formed (Figure 4A) [120]. These inclusions were also positive for ubiquitin, an autophagy substrate, p62, and thioflavin. Filamentous structures in the sarkosyl-insoluble fraction extracted from the seeded SH-SY5Y cells were labeled with pS129 and C-terminal α-syn antibodies (Figure 4A). Immuno-EM of the seeded cells showed that the α-syn inclusions consisted of filaments approximately 10 nm in diameter (Figure 4B, C). PS129-positive α-syn filaments were randomly oriented in the cytoplasm, formed reticular structures, and frequently were intermingled with mitochondria (Figure 4B, C). This presence of mitochondria in clustered α-syn filaments is a characteristic of LBs. Moreover, the introduction of recombinant α-syn filaments labeled with gold nanoparticles into SH-SY5Y cells resulted in polar filament elongation [123].
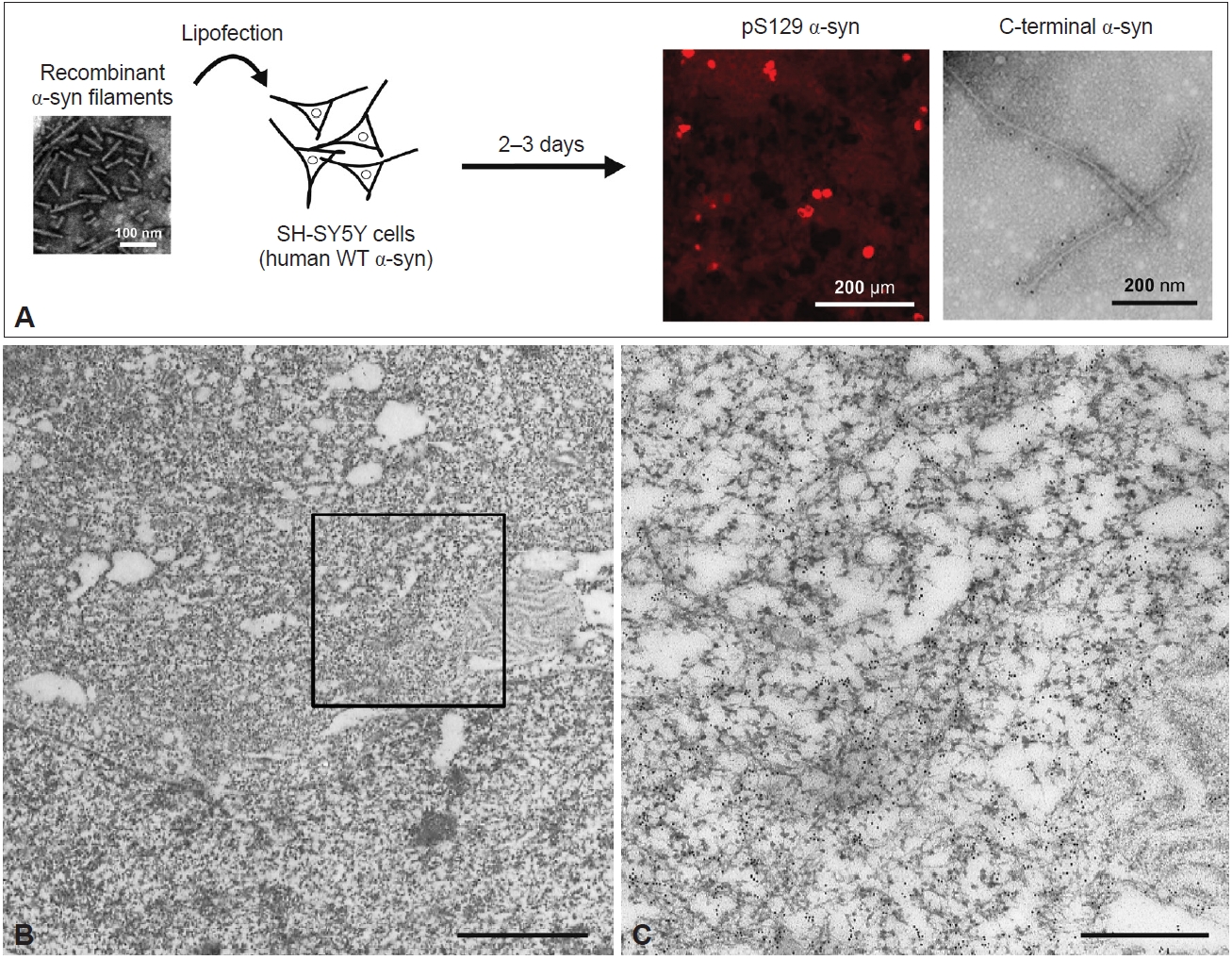
Ultrastructures of α-syn filaments observed in a cellular model of seeded α-syn aggregation. A: Schematic representation of seeded α-syn aggregation in SH-SY5Y cells. Sonicated recombinant α-syn filaments are introduced into SH-SY5Y cells by lipofection. A few days after introduction, anti-pS129-positive α-syn inclusions were detected. The 5–10 nm wide filaments extracted from the seeded cells were labeled with a C-terminal α-syn antibody (syn102). B: Immuno-EM of anti-pS129-positive α-syn filament structures in SH-SY5Y cells treated with recombinant α-syn filaments, detected with gold nanoparticles. Scale bar, 2 μm. C: Magnified image of the square marked in B. Scale bar, 500 nm. All data are original for this review. WT, wild-type; EM, electron microscopy.
Moreover, the addition of recombinant α-syn filaments to mouse primary neurons causes multiple types of insoluble α-syn pathology in a time-course-dependent manner [121]. On Day 7 after the seed treatment, small puncta and LN-like α-syn pathology with various lengths were observed, mainly in neurites, and immuno-EM revealed randomly organized or parallel-bundled pS129-positive α-syn filaments. Furthermore, larger α-syn inclusions were observed in the cytoplasm on Day 14, and some of these inclusions had an LB-like dense, round morphology. The seeded neurons at this time point showed abundant filamentous structures 14–16 nm in diameter in the cytoplasm, presynaptic terminals, postsynaptic terminals, and neurites. The interaction of α-syn filaments with organelles is suggested to occur along with the formation of cytoplasmic α-syn inclusions. Additional incubation led to the observation of LB-like α-syn inclusions distinguished by membranes on Day 21; these inclusions were composed not only of α-syn filaments but also of mitochondria, autophagosomes, and endolysosomal vesicles [124]. Immunocytochemistry showed that these LB-like α-syn inclusions were positive for an outer mitochondrial membrane protein, Tom 20, and a lysosome-associated membrane protein, LAMP1, and were partially colocalized with endoplasmic reticulum (ER) proteins. Cryo-electron tomography (ET) of the seeded rat primary neurons also revealed α-syn inclusions composed of abundant filaments, in which various organelles, such as ER, mitochondria, autophagolysosomal structures, and vesicles, were present [123]. Regarding cell-to-cell transmission, immuno-EM study of seeded human astrocytes showed that labeled recombinant α-syn filaments were observed in macropinosomes shortly after seed treatment, and some of them were transferred to neighboring naïve cells [125].
Patient-derived α-syn filaments also cause the formation of pS129-positive filamentous inclusions in cultured cells and primary cultures. Remarkably, LBD-α-syn and MSA-α-syn exhibit distinct prion-like seeding properties. The seeding efficiency of LBD-α-syn is much less than that of recombinant α-syn filaments, while MSA-α-syn induces a greater extent of seeded aggregation, which is independent of the α-syn concentration in the seed fraction [71]. The unique seeding properties of patient-derived α-syn filaments are commonly observed in various types of cultured cells and in both primary neurons and oligodendrocytes, suggesting that distinct conformations of α-syn filaments differ in their ability to recruit normal α-syn [71,73,126,127]. Cryo-ET study showed that the addition of recombinant α-syn filaments or MSA-α-syn to rat primary neurons resulted in the formation of seeded α-syn filaments with different biophysical properties [123].
These cellular studies demonstrated the formation of filamentous α-syn inclusions containing various organelles in the neuronal environment, akin to those of LBs observed in the patient’s brain. However, it remains unclear whether α-syn filaments formed in the cellular systems inherit the conformation of the original seeds. Cryo-EM structures of tau filaments extracted from SH-SY5Y cells seeded with patient-derived tau filaments appeared consistent with the possibility of templated tau filament amplification [128]. A similar approach is expected to elucidate the structure of cellular model-derived α-syn filaments and the contribution of the cellular environment to conformational templating.
α-SYN FILAMENTS IN ANIMAL MODELS
Inoculation of recombinant and patient-derived α-syn filaments into the brains and peripheral tissues of rodents and nonhuman primates induces the formation of α-syn pathology followed by intracerebral spreading [129-137]. Although the distribution of spreading varies depending on the inoculation site [138-141], pS129- and thioflavin-positive neuronal α-syn pathology is observed in inoculated animal brains and peripheral tissues (Figure 5A). As in the cellular models, MSA-α-syn exhibits a greater ability to induce α-syn pathology than LBD-α-syn [71,73,142]. However, oligodendroglial α-syn pathology is not triggered even by the inoculation of MSA-α-syn. In the M83 transgenic (Tg) mouse line expressing human A53T α-syn, heterozygous mice inoculated with MSA-α-syn developed Gallyas-silver-negative neuronal α-syn pathology, which differs from the characteristics of GCIs [143,144]. This can be explained by the very low level of α-syn expression in oligodendrocytes. Inoculation of LBD-α-syn and MSA-α-syn into KOM2 Tg mice, which express α-syn in oligodendrocytes but not in neurons, resulted in the formation of oligodendroglial α-syn pathology [73]. This, together with the lack of α-syn pathology in seed-inoculated α-syn-knockout mice [130,131], supports the structural conversion of α-syn expressed in animals to a filament form. Recombinant α-syn filament strains have been reported to induce various types of neuronal α-syn pathology, heterogeneity of disease progression and cell death in rodent brains [90,145-147].
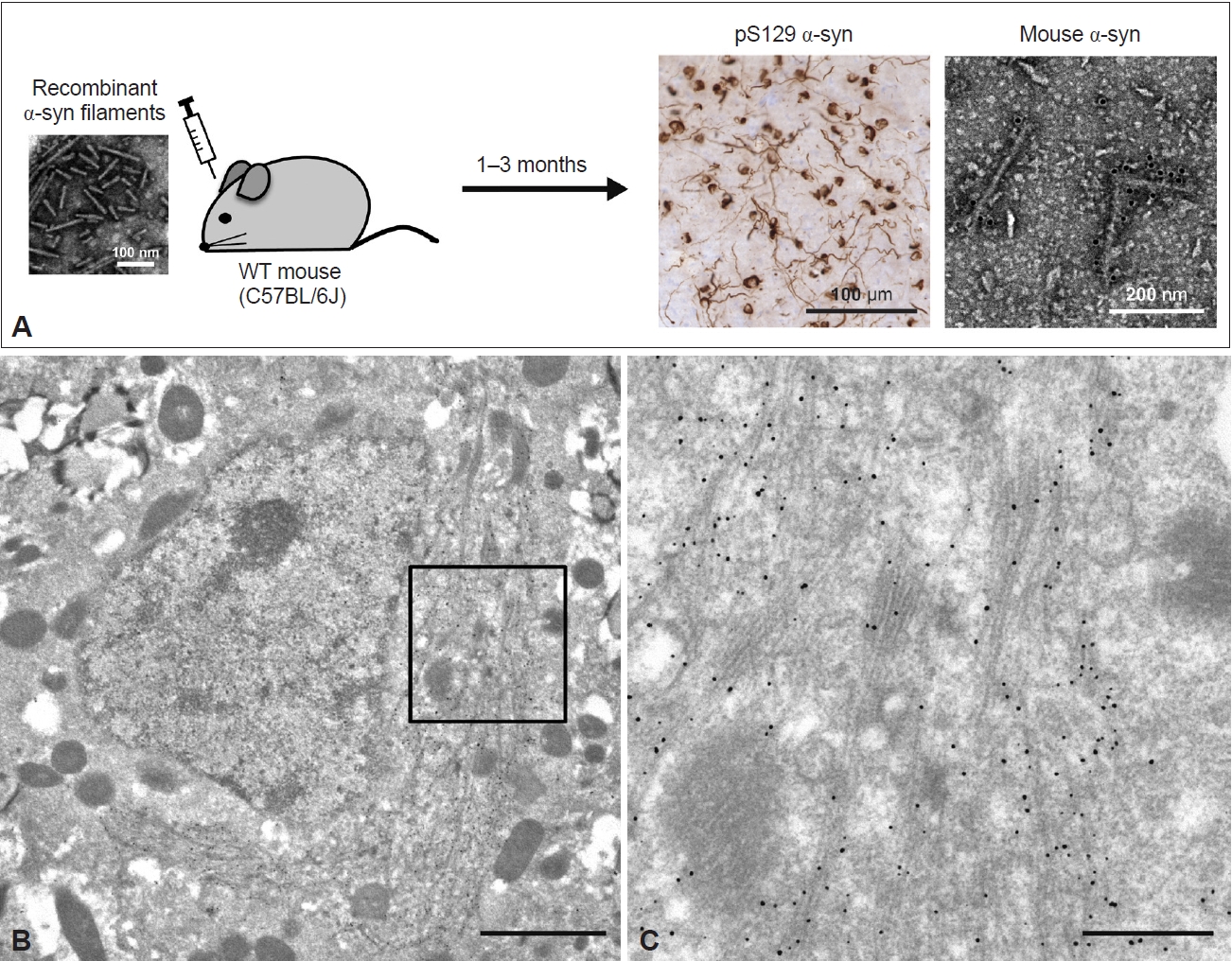
Ultrastructures of α-syn filaments observed in a mouse model of seeded α-syn aggregation. A: Schematic representation of seeded α-syn aggregation in wild-type (WT) mouse brains. Sonicated recombinant α-syn filaments were inoculated into the right striatum of WT mice. At 1–3 months after inoculation, abundant anti-pS129-positive α-syn pathology was observed. The 5–10 nm wide filaments extracted from the inoculated mouse brain were labeled with a mouse α-syn-specific antibody. B: Immuno-EM of anti-pS129-positive α-syn filament structures in the brain of WT mice at 3 months after inoculation of recombinant α-syn filaments, detected with gold nanoparticles. Scale bar, 2 μm. C: Magnified image of the square marked in B. Scale bar, 500 nm. All data are original for this review. EM, electron microscopy.
Anti-α-syn-labeled filaments 10–16 nm in diameter were randomly present in the brain and spinal cord sections of M83 homozygous mice exhibiting α-syn pathology [148]. Detergent-insoluble fractions extracted from diseased M83 homozygous and seed-inoculated or seed-administered M83 heterozygous mice contained pS129-positive α-syn filaments [149,150]. Filamentous structures 5–10 nm in diameter were also observed in the sarkosyl-insoluble fraction extracted from WT mice inoculated with recombinant human α-syn filaments (Figure 5A) [151]. These filaments were recognized by mouse α-syn-specific and pS129 antibodies, indicating that recombinant human α-syn filaments recruited endogenous mouse α-syn into filaments. Immuno-EM of the inoculated WT mouse brain sections also showed the accumulation of numerous pS129-labeled elongated α-syn filaments 10–20 nm in diameter around the nucleus (Figure 5B, C). The α-syn filaments were present randomly or in parallel, partially as bundles, and intermingled with mitochondria and some types of vesicles (Figure 5B, C). This phenomenon resembles the development of α-syn pathology observed in LBD brains and in neuronal cellular models. While the cryo-EM structures of amyloid-β and tau filaments derived from Tg and knock-in mice have already been reported [152-154], the α-syn filaments derived from animal models have not yet been precisely characterized.
IMPLICATIONS OF CONFORMATIONAL DIFFERENCES BETWEEN PATIENT- AND EXPERIMENTAL MODEL-DERIVED α-SYN FILAMENTS
As we described above, the atomic structures of α-syn filaments extracted from synucleinopathy brains are not identical to those of α-syn filaments formed in vitro. What, other than mutations in α-syn, might cause these conformational differences? First, in vitro models do not fully mimic the cellular environment. The formation of distinct types of recombinant α-syn filaments under various conditions suggests that the initial misfolding of α-syn is easily influenced by external environmental factors [96,97,155]. Also, some additional densities identified in patient-derived α-syn and tau filaments have been speculated to be solvent molecules [58,156]. Since the initial misfolding form is supposed to be inherited in the subsequent templated amplification process, it is crucial to identify the cellular environmental factors and molecules involved in the formation of disease-associated α-syn folds. The presence of lipids in LBs suggests that some nonproteinaceous cofactors may affect not only filament formation but also the core structur [61,63]. LC‒MS/MS analysis revealed 296 proteins, including synaptic proteins, in LBs, indicating that protein-protein interactions would be important for LB formation [157]. Furthermore, the cell type-specific cellular environments of neurons and oligodendrocytes may contribute to the disease-specific α-syn folds. The failure to reproduce MSA filaments in vitro has suggested that oligodendroglial environment-derived factors may be essential not only in the initial misfolding but also in the templated amplification [111]. Diverse transcriptomic profiles have been reported between cell types and brain regions [158-160], suggesting the involvement of genetic factors and their changes with aging in cell type-specific environments. However, there is a critical question to be resolved in considering the importance of the oligodendroglial environment for the formation of the MSA folds. Although the α-syn expression level in oligodendrocytes has been reported to be increased in MSA brains [161], the actual expression level and the origin of α-syn constituting GCIs remain debatable [162]. If α-syn is transferred from neurons, what is the molecular species of the transferred α-syn? The obvious structural differences between LBD and MSA filaments militate against the possibility that filamentous α-syn is transferred from neurons to oligodendrocytes [48,58], instead favoring the transfer of monomeric or nonfilamentous oligomeric α-syn. Some types of cellular stress and neuronal impairment in the affected neurons may cause an increase in the SNCA mRNA level in oligodendrocytes and/or accelerate GCI formation. The experimental models mimicking neuronal α-syn pathology showed similar characteristics to LBs in LBD brains regarding the copresence of organelles, vesicles, and membrane structures with elongated α-syn filaments, which were also observed in neurons treated with MSA-α-syn [120,123,124,151]. These observations indicate that this is a hallmark of α-syn pathogenesis in neurons rather than a consequence of the type of α-syn seeds. Further studies are needed to elucidate in detail the α-syn pathogenesis in oligodendrocytes.
In addition, the absence of post-translational modifications (PTMs) in in vitro models may influence the filament core structure. The additional densities in patient-derived α-syn filaments display similar biochemical characteristics [59], and PTMs might be involved in them. C-terminal phosphorylation modifies the negative charge of normal α-syn, increases the exposure of the N-terminal structure, and changes its interaction with environmental factors [163]. Phosphorylation at Y39 causes the formation of an electrostatic interaction network at the N-terminal region and changes the filament core structure, supporting the potential involvement of PTMs in α-syn filament formation [101]. Lysine PTMs are located on the outside of the structured filament core in synucleinopathy brains [48], suggesting that these modifications occur after the filament core is formed. However, the possible contributions of acetylation, ubiquitination and other PTMs need to be further investigated, and the PTM profiles of LBD and MSA should also be established in more detail. Immunoblot analysis of sarkoayl-insoluble α-syn derived from LBD and MSA brains show distinct high-molecular α-syn bands [71,164,165], indicating disease specificity in the PTM profile. In the case of tau, N-/C-terminal truncation has been shown to be essential for the in vitro formation of the folds seen in AD and chronic traumatic encephalopathy [156,166]. Disease-specific PK-resistant α-syn bands in LBD and MSA appear to be consistent with different C-terminal truncation processes [73,167].
CONCLUSIONS
The ultrastructural and biochemical properties of pathological α-syn accumulated in synucleinopathy brains have been extensively characterized. Diseases in the same pathological category share structurally and biochemically identical α-syn filaments, implying that the conformation of α-syn filaments contributes to the pathological diversity of synucleinopathy. Cryo-EM studies of patient-derived α-syn filaments are consistent with prion-like, templated amplification of α-syn filaments in the human brain. However, the mechanisms by which α-syn adopts filamentstructural polymorphisms remain unclear. Conformational differences in α-syn filaments derived from patients’ brains and from experimental models indicate that environmental factors and molecules are involved in the formation of patient-derived α-syn folds, and these remain to be identified. More detailed biochemical characterization of LBD-α-syn and MSA-α-syn may be helpful in this regard. Recently, heterogeneity of in vitro and in vivo seeding properties triggered by α-syn seeds derived from patients with a single disease has been reported, suggesting a potential association with clinical phenotypes [113,168-171]. Although it is unknown whether there are structurally distinguishable subtypes within LBD or MSA, establishing links between seeding properties and other factors, including disease duration and progression, cognitive, motor and psychiatric symptoms and histopathological features, would further advance SAA-based early diagnosis. In addition, the patient-derived α-syn filament folds are expected to be helpful for positron emission tomography (PET) imaging and for developing therapeutic strategies. The atomic structure of recombinant α-syn filaments bound with a PET probe, [18F]-F0502B, has been reported [172], and the development of PET probes targeting the disease-specific filament core might enable discrimination between LBD and MSA at an early disease stage [173]. Similarly, the development of structure-based aggregation inhibitors and antibody therapies seems to be a promising avenue for future studies [174,175].
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
This work was supported by Japan Science and Technology Agency, CREST Grant Number JPMJCR18H3 (to M.H.), Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP18ek0109391, JP-18dm0207019 (to M.H.), JSPS KAKENHI Grant Numbers JP20K16482 (to A.T.).
Author Contributions
Conceptualization: Airi Tarutani, Masato Hasegawa. Funding acquisition: Airi Tarutani, Masato Hasegawa. Investigation: Airi Tarutani, Masato Hasegawa. Writing—original draft: Airi Tarutani. Writing—review & editing: Masato Hasegawa.
Acknowledgements
We are grateful to the patients and their families for making tissues available for research. Tissue samples used for immunohistochemistry and immuno-EM were supplied by the Japan Brain Banks at the Tokyo Metropolitan Institute of Geriatrics and Gerontology and Sagamihara National Hospital. Molecular graphics in Figure 3B were created with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.