Diagnosis and Clinical Features in Autoimmune-Mediated Movement Disorders
Article information

Abstract
Movement disorders are common manifestations in autoimmune-mediated encephalitis. This group of diseases is suspected to be triggered by infection or neoplasm. Certain phenotypes correlate with specific autoantibody-related neurological disorders, such as orofacial-lingual dyskinesia with N-methyl-D-aspartate receptor encephalitis and faciobrachial dystonic seizures with leucine-rich glioma-inactivated protein 1 encephalitis. Early diagnosis and treatment, especially for autoantibodies targeting neuronal surface antigens, can improve prognosis. In contrast, the presence of autoantibodies against intracellular neuronal agents warrants screening for underlying malignancy. However, early clinical diagnosis is challenging because these diseases can be misdiagnosed. In this article, we review the distinctive clinical phenotypes, magnetic resonance imaging findings, and current treatment options for autoimmune-mediated encephalitis.
INTRODUCTION
Autoimmune-mediated encephalitis has heterogeneous presentations, and an increasing number of autoantibodies have been discovered. Movement disorders are one of the most common features in these kinds of diseases. Certain movement disorders can correlate with specific autoantibodies. Most are also comorbid with limbic encephalitis, epilepsy, or peripheral neuropathy. Diagnosing these disorders as early as possible is crucial because some antibodies are associated with occult neoplasia, which is potentially treatable. Delayed diagnosis may have life-threatening consequences or cause permanent morbidity [1,2]. However, autoantibodies can cause many overlapping presentations, and variable presentations can occur with each autoantibody. Therefore, clinical diagnosis is challenging.
In this review, we summarize the distinctive phenotypes, clinical course, tumor association, imaging investigation, and laboratory examination for autoimmune-mediated encephalitis. This serves as a practical guide for differential diagnosis as well as the possible treatment options for the spectrum of diseases.
CLASSIFICATION OF AUTOANTIBODIES AND PATHOPHYSIOLOGICAL MECHANISMS
Autoantibodies are distinguished by their different immunological mechanisms and fall into three groups: neuronal surface antibodies, antibodies targeting intracellular synaptic proteins, and antibodies targeting cytoplasmic and nuclear antigens. These autoantibodies can trigger T-cell or B-cell immune reactions (Table 1) [3,4].
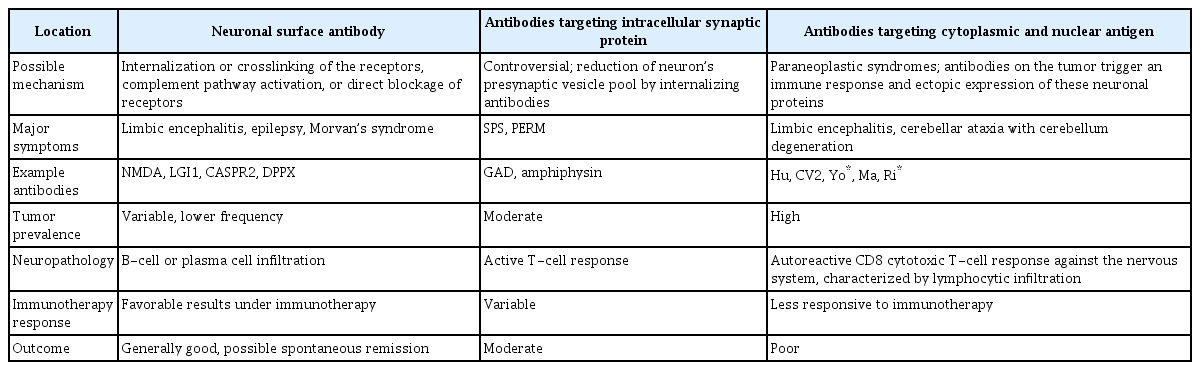
Autoantibody-associated central nervous system symptoms
The first group of antibodies is neuronal surface antibodies, which are correlated less strongly with malignancy than are intracellular antibodies. Autoantibodies have been found to be produced from B cells induced by antigens from tumor cells or viruses [5,6]. The autoantibodies target surface receptors and internalize target receptors or influence protein–protein interactions, causing secondary receptor dysfunction [4,7,8]. The antibodies and receptor effects are considered to be reversible. The possible pathophysiology of N-methyl-D-aspartate (NMDA) receptor encephalitis may be caused by autoantibodies targeting neuron surface antigens. The alteration of the receptor that may induce internalized NMDA receptors causes NMDA receptor hypofunction. The interneuron in pyramidal cells may change the nucleus accumbens (NAc) activity. Increasing the production of dopamine causes limbic encephalitis and dyskinesia (Figure 1) [5,9-11]. Most patients present with seizures, cognitive dysfunction, and variable movement disorders, such as orolingual dyskinesia, ataxia, and parkinsonism [4,12-15]. Furthermore, these movement disorders are thought to have a favorable prognosis with early immunological treatment as well as adequate tumor removal [12,15,16].
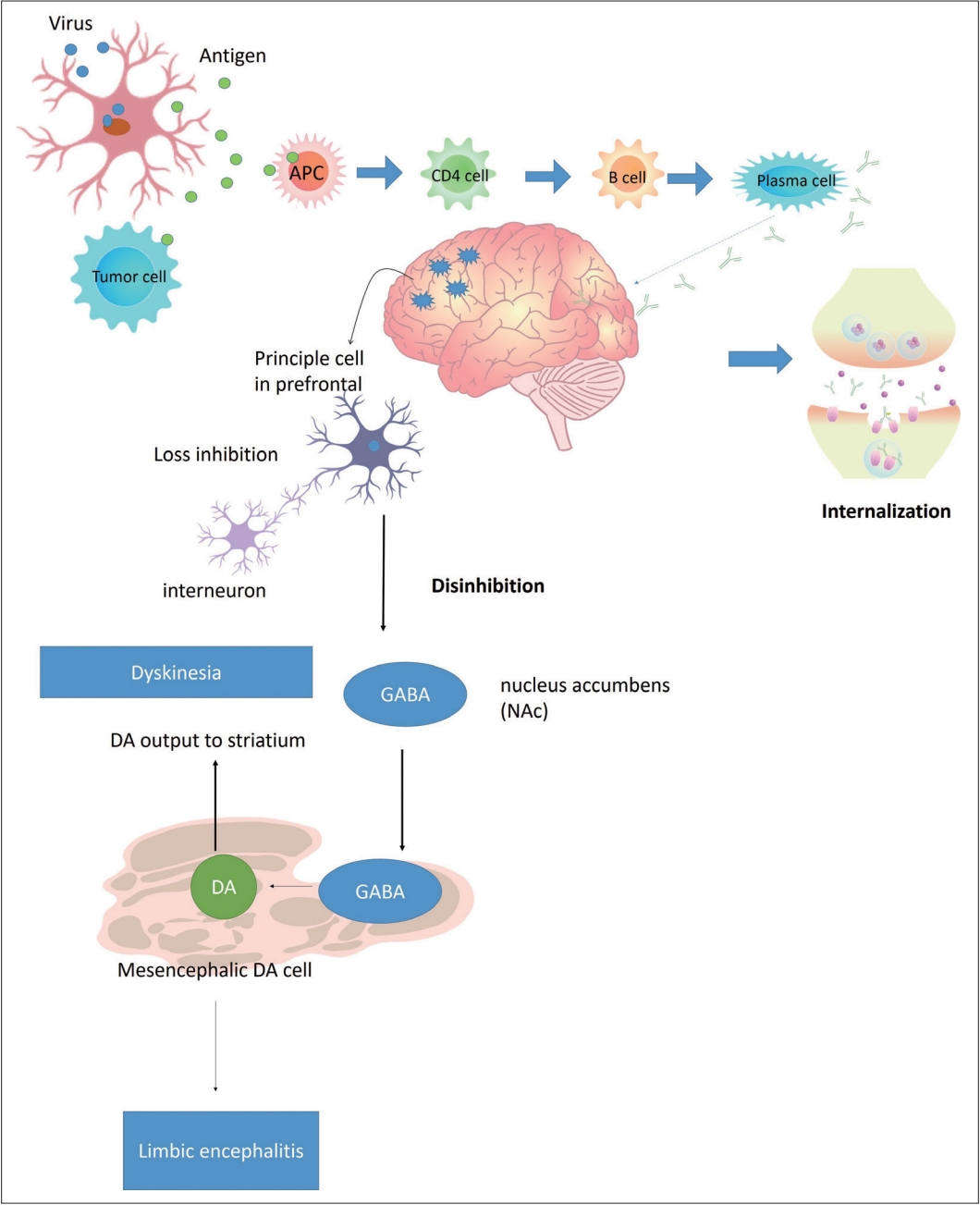
Possible pathophysiology of NMDA receptor encephalitis. The possible pathophysiology of NMDA receptor encephalitis may be caused by autoantibodies targeting neuron surface antigens. The alteration of the receptor may induce internalization of NMDA receptors, which is related to NMDA receptor hypofunction. The interneuron in pyramidal cells may change NAc activity. In terms, increasing the production of DA in the striatum and dorsal lateral prefrontal cortex may be related to limbic encephalitis and dyskinesia. APC, antigen-presenting cells; GABA, gamma-aminobutyric acid; NAc, nucleus accumbens; DA, dopamine; NMDA, N-methyl-D-aspartate.
The second group of antibodies targets intracellular synaptic proteins and is primarily associated with stiff-person syndrome (SPS) and ataxia [17,18]. Typically, the target protein antigens are located in the intracellular space of the synapse and prompt the vesicle pool to release an inhibition signal [17,19]. Uncertainty remains regarding the pathophysiology, but both T-cell and Bcell involvement is suspected [20]. Some reports have shown intrathecal synthesis and neuron internalization of anti-glutamic acid decarboxylase antibody (anti-GAD Ab) [21,22]; another study revealed that GAD65-reactive CD4 T-cells may produce interferon-gamma (INF-γ) [23]. GAD65 is one of two enzymes that catalyze the formation of the major neuroinhibitor gamma-aminobutyric acid (GABA). Anti-GAD Ab disturbs synaptic vesicles and then decreases the secretion of GABA. The reduction in GABA levels may induce an increase in glutamate due to a lower inhibition signal. Glutamate may further activate microglia and reduce the reuptake of glutamate by impairing excitatory amino acid transporters (EAATs). The increase in glutamate concentration may induce stimulation of neuronal nitric oxide synthase and calpain I, leading to mitochondrial dysfunction and cell apoptosis (Figure 2) [24,25]. The possible pathophysiological mechanism related to ataxia is an imbalance between GABA and glutamate, which causes excitotoxicity to neuronal cells [25,26]. The association with malignancy differs among each antibody, and the prognosis for disorders associated with this group of antibodies is more favorable than that for anticytoplasmic and nuclear antigen antibody disorders; however, these antibodies are more refractory than neuronal surface antibodies [12].
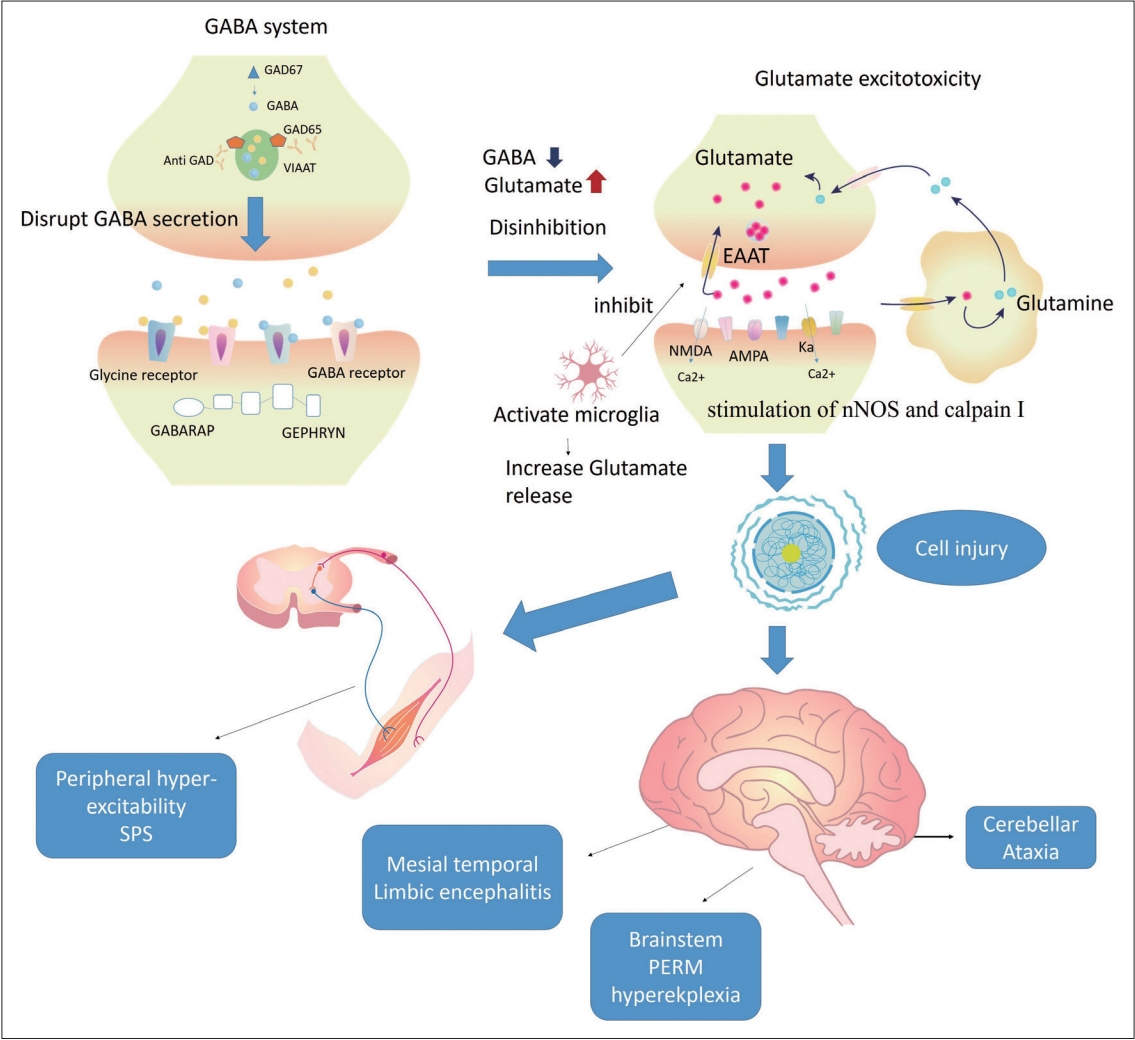
Possible pathophysiology of anti-GAD antibody-related disorders. The possible pathophysiological mechanism is an imbalance between GABA and glutamate, which causes excitotoxicity to neuronal cells. Anti-GAD Ab internalized and disturbed the synaptic vesicles, GAD65, and secreted GABA. The reduction of GABA levels may induce an increase in glutamate due to a lower inhibition signal. Glutamate may further activate microglia, leading to increased glutamate release and reduced reuptake of glutamate by impaired EAATs. The increase in glutamate concentration may induce stimulation of nNOS and calpain I, leading to mitochondrial dysfunction and cell apoptosis. A decrease in GABA may cause hyperexcitability in the peripheral or central nervous system. GABA, gamma-aminobutyric acid; GAD, glutamic acid decarboxylase; VIAAT, vesicular inhibitory amino acid transporter; GABARAP, gamma-aminobutyric acid receptor-associated protein; anti-GAD ab, anti-glutamic acid decarboxylase antibody; EAAT, excitatory amino acid transporter, NMDA, N-methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate; nNOS, neuronal nitric oxide synthase; SPS, stiff-person syndrome, PERM, progressive encephalomyelitis with rigidity and myoclonus.
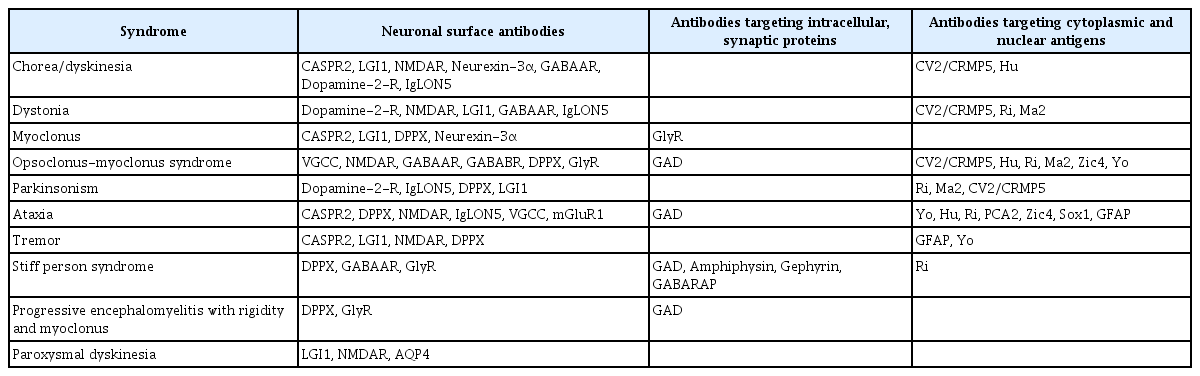
The final group of antibodies, which target cytoplasmic and nuclear antigens, are also known as onconeural antibodies and are associated with several paraneoplastic neurological syndromes [27-30]. These syndromes most often present as limbic encephalitis, which may be accompanied by subacute cerebellar ataxia, chorea, and parkinsonism (Table 2) [12,29,31]. The pathophysiological mechanism for this group of diseases is that onconeural antigen-specific CD4 T-cells may recruit tumor antigen-specific cytotoxic CD8 T cells and activate plasma cells to produce onconeural antibodies (Figure 3) [32,33]. These onconeural antigen-specific T cells cross the blood brain barrier and reach the central nervous system. Then, the intracellular antigen upregulates major histocompatibility complex class 1, which causes a cytotoxic CD8 T-cell misdirected response against the nervous system, which induces variable manifestation [3,4,29]. These autoantibodies may not be the major cause of neuronal damage, but they are a potential biomarker for this group of diseases [3]. Pathology findings have indicated lymphocyte infiltration with extensive neuron degeneration, which is considered to be primarily involved with pathogenic cytotoxic T cells [3,8,34]. Most neurological symptoms of encephalitis related to this group of autoantibodies are highly associated with malignancy as well as a poor response to immunotherapy [29,34].
Movement disorders associated with autoantibodies
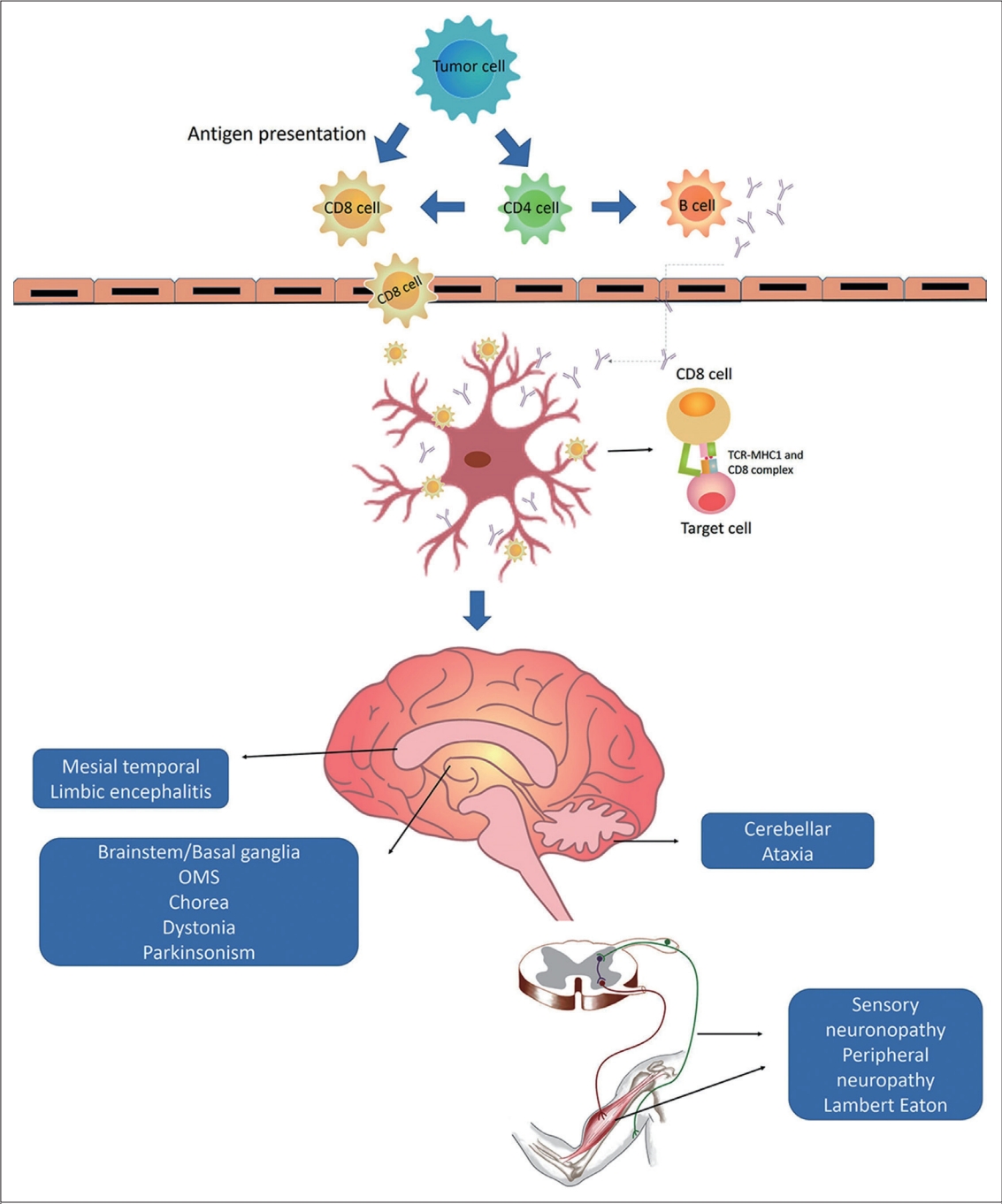
Possible mechanism in onconeural antibody-related disorders. Onconeural antigens are expressed in tumor cells. Onconeural antigen-specific CD4 T cells may recruit tumor antigen-specific cytotoxic CD8 T cells and activate plasma cells to produce onconeural antibodies. These onconeural antigen-specific T cells cross the blood brain barrier and reach the central nervous system. Then, the intracellular antigen upregulates MHC class 1, which causes a cytotoxic CD8 T-cell misdirected response against the nervous system and induces variable disorders. MHC, major histocompatibility complex; TCR, T cell receptor; OMS, opsoclonus-myoclonus syndrome.
CLINICAL APPROACH AND DIAGNOSIS
The diagnosis of autoimmune-mediated autoantibodies is challenging in clinical practice. A careful history assessment may be helpful. The clinical courses of these disorders usually involve subacute onsets of cognitive change or psychiatric presentation [15,35]. If patients present with variable movement manifestations, especially combined movement disorders, neuronal surface antibodies are more likely. Moreover, if patients present with SPS, intracellular synaptic protein antibodies are the first to be considered. In contrast, if a patient’s major symptom is subacute onset ataxia and suspected opsoclonus-myoclonus syndrome, clinicians may consider antibodies targeting intracellular nuclear antigens (Supplementary Table 1 in the online-Data Supplement).
Cerebral spinal fluid (CSF) may exhibit pleocytosis, which could be misdiagnosed as virus-related encephalitis. Electroencephalograms (EEGs) often reveal diffuse slow wave activity, epileptiform discharge, or even status epilepticus [4,6,36]. Brain magnetic resonance imaging (MRI) findings are typically normal, but they may show hyperintense unilateral or bilateral T2/fluid-attenuated inversion recovery (FLAIR) signals in the mesial temporal lobes and restricted diffusion relative to normal [4,13]. In addition, fluorine-18 deoxyglucose-positron emission tomography (18F-FDG-PET) scans may play a crucial role in diagnosis. 18F FDG-PET may be more sensitive than brain MRI, and the presence of some antibodies may be associated with specific metabolic patterns [37-39].
TREATMENT PRINCIPLES
Symptomatic therapy for involuntary movement should be provided for most patients. Of patients who have severe disease, 87% present movement disorders [3]. Metabolic problems such as infection, electrolyte imbalance or pain may exacerbate hyperkinetic movement disorders. Acute respiratory failure or rhabdomyolysis may be induced by uncontrolled dystonia or dyskinesia. Deep sedative medication should be used for refractory movement disorders or status epilepticus [3,34].
Immunotherapy must be initiated as soon as possible. The first-line therapy is intravenous (IV) methylprednisolone at a dosage of 1 g for 3–5 days, which has efficacy due to wide function for immunosuppression as well as T-cell depletion, followed by oral prednisolone; IV immunoglobulin (IVIg) at a dosage of 0.4 g/kg for 5 days; and/or plasmapheresis [4,8,30]. If patients are highly suspected of autoimmune-mediated encephalitis by classical onconeuronal antibodies, methylprednisolone or other T-cell-directed therapies are preferred options over IVIg or plasmapheresis [3,35,36]. However, patients with classical onconeuronal antibody-related encephalitis may show only modest effects on immunosuppression and tend to respond best to cancer therapy [3,35]. Combined therapy is suggested if patients have a severe initial presentation, such as new onset refractory status epilepticus, or fail to respond to the initial agent [3,13].
If the patient is refractory to first-line treatment, second-line therapy should be considered after 2–3 weeks [40,41]. Clinicians should consider rituximab anti-CD20 therapy for 4 weeks with or without IV cyclophosphamide for 3–6 months [30,37-39]. A common regimen of rituximab is 375 mg/m2 weekly for 4 weeks. In addition, the common regimen for cyclophosphamide is 750 mg/m2 for 3–6 months [3]. If the patient still responds poorly, alternative treatment should be considered, such as interleukin (IL)-6 inhibition (tocilizumab), low-dose IL-2, or bortezomib [30,38-40,42]. Recently, a Janus kinase inhibitor, tofacitinib, may be a new option if patients are refractory to second-line therapy [41,42]. In addition, careful examination for underlying malignancy is essential. Tumor removal is crucial in patients with any type of autoimmune-mediated encephalitis. Patients may exhibit some improvement after tumor ablation. Long-term maintenance therapy, such as low-dose prednisolone and steroid-sparing medication, should be considered for patients whose diseases relapse or who respond poorly to medication (Figure 4) [38,39].
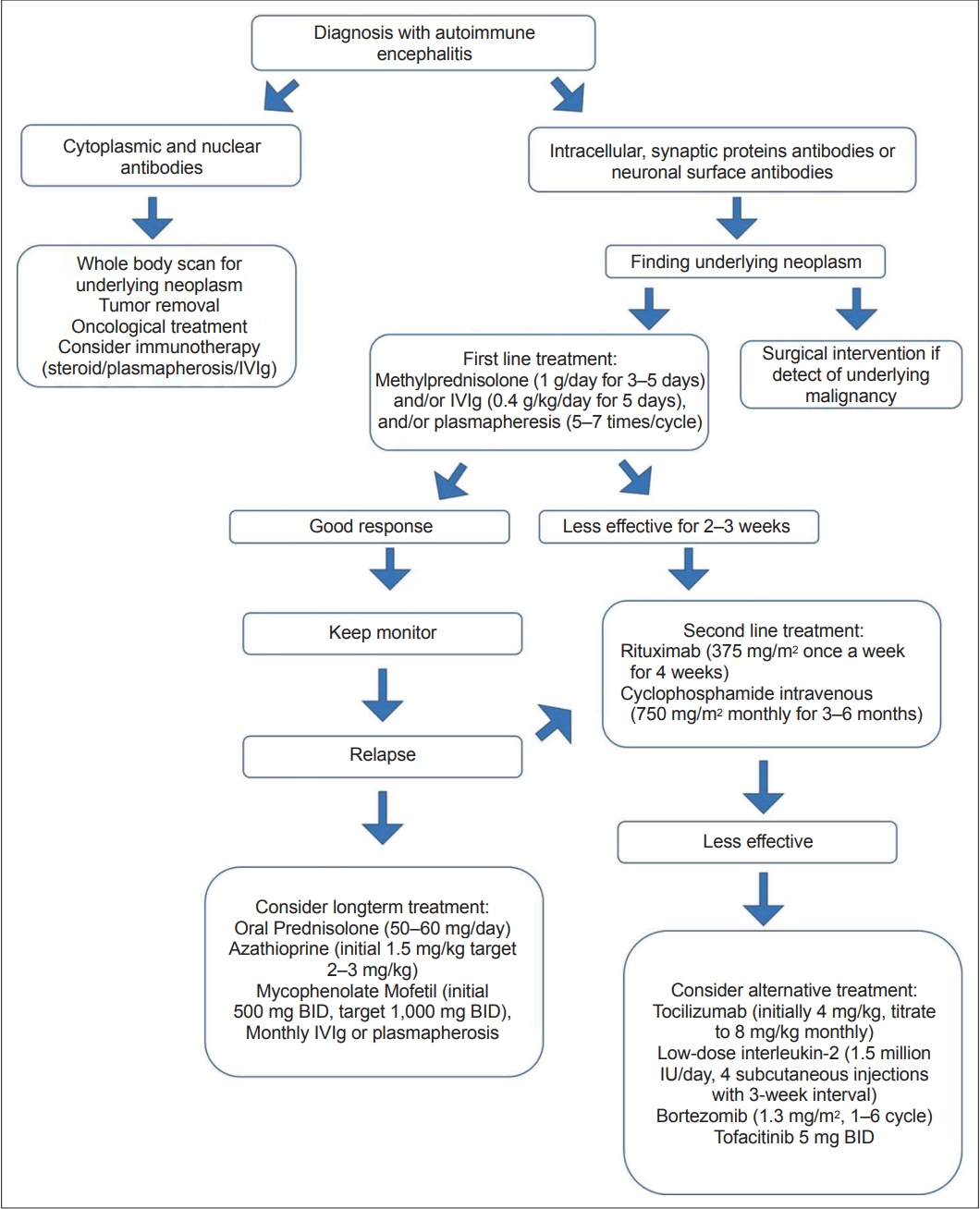
Treatment principle for autoimmune-mediated encephalitis. IVIg, intravenous immunoglobulin.
MOVEMENT DISORDERS ASSOCIATED WITH ANTI-NEURONAL SURFACE PROTEIN ANTIBODIES
Clinical presentation and diagnosis of anti-N-methyl-D-aspartate receptor encephalitis
Anti-NMDA receptor encephalitis is a well-known type of autoimmune-mediated encephalitis. It typically affects young women between the ages of 16 and 42 years, and 25%–50% of cases present with ovarian teratoma [1,36]. Nontumor cases are usually observed in male patients or very young girls [36,43]. The disease course generally seems to follow the same pattern [1,36]. In the prodromal stage, 70% of patients experience headaches, fever, and upper respiratory infection symptoms. Following this stage, agitation, psychiatric symptoms, catatonia, hallucinations, new-onset seizures, and speech and memory impairment can be observed. After weeks to months, orolingual-facial dyskinesia is the most characteristic feature in anti-NMDA receptor encephalitis, accompanied by dystonia, chorea, myoclonus, stereotypic movement, ataxia, and parkinsonism over the trunk and all extremities (Supplementary Table 2 in the online-only Data Supplement) [1,36,43,44]. Clinicians should be aware of these combined movement disorders, which may cause self-injury. Overall, the most common movement disorders are dystonia, stereotypies, and chorea [4,43,45]. Tetrabenazine, clonazepam, or botulinum injection might provide some symptomatic control. Some patients may present with hyperkinetic crises, which warrant the use of intensive sedative medication. In addition, decreased levels of consciousness, dysautonomia, and central hypoventilation may increase the patient’s risk of mortality [44].
CSF analyses have revealed a rate of lymphocyte pleocytosis of 68%–98%, normal to mildly elevated protein levels, and oligoclonal bands (OCB) in 50%–60% of patients [36,46-48]. Anti-NMDA receptor antibodies can be found in CSF with a higher sensitivity than in serum, and a higher titer may yield a poorer outcome [49]. Abnormal EEG findings were observed in 80% of patients [50]. The most characteristic presentation in anti-NMDA receptor encephalitis is extreme delta brush, which is associated with poor recovery [51,52]. Brain MRI revealed unremarkable findings in half of patients, and half of patients may exhibit T2/FLAIR signal hyperintensity in the hippocampus, temporal cortex, frontal cortex, and brainstem [53]. FDG-PET has revealed that medial occipital lobe hypometabolism may be an early biomarker and may correlate with improving neurologic status [37,38,53].
Clinical presentation and diagnosis of leucine-rich glioma inactivated protein 1 antibody–related syndromes
Leucine-rich glioma inactivated protein 1 (LGI1) antibodies are voltage-gated potassium channel (VGKC)-associated proteins. They usually affect middle-aged male patients [54]. Less than 20% of cases are associated with tumors, which are usually thymoma [13,55]. The common initial symptoms are amnesia and seizures. The most distinctive feature is faciobrachial dystonic seizures (FBDS), which in some patients may be combined with focal seizures or general tonic–clonic seizures. FBDS typically occur in clusters 2–3 weeks before cognitive impairment, followed by personality change and psychosis. Sleep disorders occur in 50% of patients [2,12,31,55,56]. Very rarely, patients present with Morvan’s syndrome. Other movement disorders, including chorea, parkinsonism, and myoclonus, are unusual [12,55].
CSF analysis may reveal normal results in 60%–75% of patients, and LGI1 antibodies are mostly detected in CSF [46]. Hyponatremia occurs in 70% of patients [54,57]. EEG may reveal epileptiform discharges or focal slow wave activity. However, most patients who present with FBDS do not exhibit epileptiform discharges [56]. Brain MRI reveals T2 hyperintensity in the mesial temporal lobe in more than half of patients [56,58]. MRI reveals basal ganglia involvement only in patients with FBDS [59]. PET scans may reveal frontal lobe hypometabolism [13]. Early immunotherapy can yield favorable outcomes and may also prevent limbic encephalitis [57].
Clinical presentation and diagnosis of contactin-associated protein-like 2 antibody–related syndromes
Contactin-associated protein-like 2 (CASPR2) antibodies are also VGKC-associated antibodies. CASPR2 antibody–related syndromes typically affect middle-aged and older male adults. Fewer than 30% of patients have associated tumors; these are usually thymoma [60], but other neoplasms have also been reported [61-63]. The core symptoms of CASPR2 antibody–related autoimmunity are Morvan’s syndrome, neuropathic pain, peripheral nerve hyperexcitability, and limbic encephalitis. Cognitive decline is observed in 80% of patients [54]. Muscle cramps, stiffness, and neuromyotonia occur because of peripheral nerve hyperexcitability [56]. This is one of the core features of Morvan’s syndrome, which suggests the need for detailed screening of the underlying thymoma [64]. Various movement disorders are observed, especially cerebellar ataxia [65]. Chorea and orthostatic myoclonus have also been reported [12,45,66].
CSF analysis may show mildly elevated protein levels or pleocytosis in 30% of patients as well as T2/FLAIR bilateral mesial temporal hyperintensity [13,60]. Electromyography (EMG) can detect neuromyotonia, fasciculation, and myokymic discharge. CASPR2 antibodies have been detected in both serum and CSF; these findings mostly revealed neuromyotonia in the serum group and epilepsy in the CSF group [55,60]. The disease is usually responsive to treatment, and patients have a fair prognosis.
MOVEMENT DISORDERS ASSOCIATED WITH ANTI-INTRACELLULAR SYNAPTIC PROTEIN ANTIBODIES
Clinical presentation and diagnosis of anti-glutamic acid decarboxylase antibody–related syndromes
Anti-GAD Ab typically affects middle-aged female patients [67]. These antibodies are often comorbid with type 1 diabetes mellitus and thyroiditis. In rare cases, they are associated with small cell lung cancer or thymoma [68]. The clinical course can involve chronic or subacute onset stiff-person spectrum disorder, cerebellar ataxia, palatal myoclonus, episodic vertigo, limbic encephalitis, and drug-resistant epilepsy [18,68,69]. Some patients present with progressive encephalomyelitis with rigidity and myoclonus [70]. CSF analysis may reveal anti-GAD Ab and OCB in 25%–67% of patients [46,71]. EMG demonstrates continuous agonist and antagonist motor activity, typically in axial muscles, and clinical observation reveals painful spasms in limb and axial muscles [19,72]. EEG may show epileptiform discharge, and brain MRI can be normal or reveal T2 FLAIR bilateral mesial temporal hyperintensity [8,17]. The antibody titers do not correlate with disease severity and treatment response [73]. First-line immunotherapy can produce a partial response [74-76]. Second-line therapy with rituximab and/or cyclophosphamide has been proposed. Patients with SPS and epilepsy may benefit from high-dose benzodiazepines combined with baclofen and anticonvulsants for symptomatic therapy. The disease outcome is variable and usually requires long-term immunosuppressive therapy [12,17,74,77].
MOVEMENT DISORDERS ASSOCIATED WITH ANTIBODIES AGAINST INTRACELLULAR ANTIGENS
Clinical presentation and diagnosis of anti-Hu antibody–related syndromes
Anti-Hu syndrome is one of the most prevalent paraneoplastic neurological disorders. It is associated with small cell lung cancer in 85% of patients and mostly affects middle-aged and older adult male patients [78]. Presentation tends to vary; the most common presentation is sensory neuronopathy, seen in more than half of patients, followed by cerebellar ataxia (10%–22% of patients), limbic encephalitis (9%–15% of patients), and brainstem encephalitis (8% of patients). Some patients with sensory neuropathy may have autonomic symptoms [78,79]. Movement disorders usually present as cerebellar ataxia [12,31]. Some patients exhibit chorea, opsoclonus-myoclonus, and pseudoathetosis related to sensory neuropathy [48]. CSF analysis findings are typically normal or show mildly elevated protein levels and pleocytosis. In addition, anti-Hu autoantibodies can be detected in CSF and serum, but the titers are not correlated with the outcome [80,81]. EEG may reveal focal or generalized epileptiform discharge as well as focal slow or normal wave activity [82]. MRI shows hyperintensity in deep gray nuclei, temporal lobe, or white matter T2/FLAIR signals [83]. FDG-PET can reveal extensive bilateral mesiotemporal hypermetabolism and help to detect underlying neoplasms [84,85]. Overall, the prognosis is poor, but some patients may improve with tumor ablation or adequate chemotherapy with or without immunotherapy [34,79].
CONCLUSION
Recognizing the warning signs of autoimmune–mediated encephalitis is crucial because some types may be treatable, especially when treatment is started early. Otherwise, long-term morbidity or mortality may occur because o1f severe complications. The underlying mechanisms of pathogenesis causing nervous system dysfunction remain unclear. Furthermore, many patients do not substantially improve with current immunotherapy. More precise medication must be developed for specific autoantibodies and groups of autoimmune-mediated encephalitis.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.21077.
Supplementary Table 1
Autoantibodies and movement disorders
Supplementary Table 2.
Antibody-associated movement disorders and tumor associations
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
This work was supported by Chang Gung Memorial Hospital, Taipei, Taiwan (BMRP 778).
Author Contributions
Conceptualization: Yih-Ru Wu, Pei-Chen Hsieh. Supervision: Yih-Ru Wu. Validation: Yih-Ru Wu. Visualization: Yih-Ru Wu. Writing—original draft: Pei-Chen Hsieh. Writing—review & editing: Pei-Chen Hsieh, Yih-Ru Wu.