 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 17(2); 2024 > Article
-
Viewpoint
A Practical Guide for Clinical Approach to Patients With Huntington’s Disease in Korea -
Chaewon Shin1,2
 , Ryul Kim3, Dallah Yoo4, Eungseok Oh2,5, Jangsup Moon6,7, Minkyeong Kim8, Jee-Young Lee3
, Ryul Kim3, Dallah Yoo4, Eungseok Oh2,5, Jangsup Moon6,7, Minkyeong Kim8, Jee-Young Lee3 , Jong-Min Kim9, Seong-Beom Koh10, Manho Kim6, Beomseok Jeon11, on behalf of the Korean Huntington’s Disease Society
, Jong-Min Kim9, Seong-Beom Koh10, Manho Kim6, Beomseok Jeon11, on behalf of the Korean Huntington’s Disease Society -
Journal of Movement Disorders 2024;17(2):138-149.
DOI: https://doi.org/10.14802/jmd.24040
Published online: March 12, 2024
1Department of Neurology, Chungnam National University Sejong Hospital, Sejong, Korea
2Department of Neurology, Chungnam National University College of Medicine, Daejeon, Korea
3Department of Neurology, SMG-SNU Boramae Medical Center, Seoul National University College of Medicine, Seoul, Korea
4Department of Neurology, Kyung Hee University Hospital, Kyung Hee University College of Medicine, Seoul, Korea
5Department of Neurology, Chungnam National University Hospital, Daejeon, Korea
6Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
7Department of Genomic Medicine, Seoul National University Hospital, Seoul, Korea
8Department of Neurology, Gyeongsang National University Hospital, Jinju, Korea
9Department of Neurology, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Seongnam, Korea
10Department of Neurology, Korea University Guro Hospital, Korea University College of Medicine, Seoul, Korea
11Department of Neurology, BJ Center for Comprehensive Parkinson Care and Rare Movement Disorders, Chung-Ang University Health Care System, Hyundae Hospital, Namyangju, Korea
- Corresponding author: Jee-Young Lee, MD, PhD Department of Neurology, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul National University College of Medicine, 20 Boramae-ro 5-gil, Dongjak-gu, Seoul 07061, Korea / Tel: +82-2-870-2476 / Fax: +82-2-831-2826 / E-mail: wieber04@snu.ac.kr
Copyright © 2024 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 729 Views
- 57 Download
- Huntington’s chorea was initially described in 1872 by Dr. George Huntington (1850–1916) [1]. Roughly a century later, in 1988, the first clinically diagnosed cases of Huntington’s disease (HD) in South Korea were reported [2]. Following the identification of the genetic cause of HD [3], the first genetic analysis of Huntington’s chorea in South Korea was published in 1996 [4].
- In South Korea, the genetic test for HD has been covered by the National Health Insurance System (NHIS) since August 2005. Advancements in genetic testing and a greater understanding of the disease have made HD diagnosis easier than in the past. A recent study examining the 10-year prevalence of HD in South Korea, based on data registered between 2010 and 2019 in the NHIS database, reported an annual incidence of 0.29/100,000 and a 10-year prevalence of approximately 2.2 per 100,000 [5].
- Despite advancements in understanding this disease, the clinical management and medical infrastructure for HD remain notably limited in South Korea. Approximately one-third of individuals diagnosed with HD discontinue medical follow-up, and among those who continue to seek medical care, there is a tendency to incur substantial medical expenses [5]. The annual medical costs for an individual with HD are estimated to be approximately 6 million KRW or more, which is sustained over a period of 9 years following diagnosis [5]. These statistics underscore the urgent need for significant improvements in various aspects of our medical system to provide effective support for patients with HD and their families in South Korea. A recent analysis on caregiver burdens of HD patients in Korea revealed that the caregiving burdens for HD patients are notably high and comparable to those expected in patients with more common dementias [6].
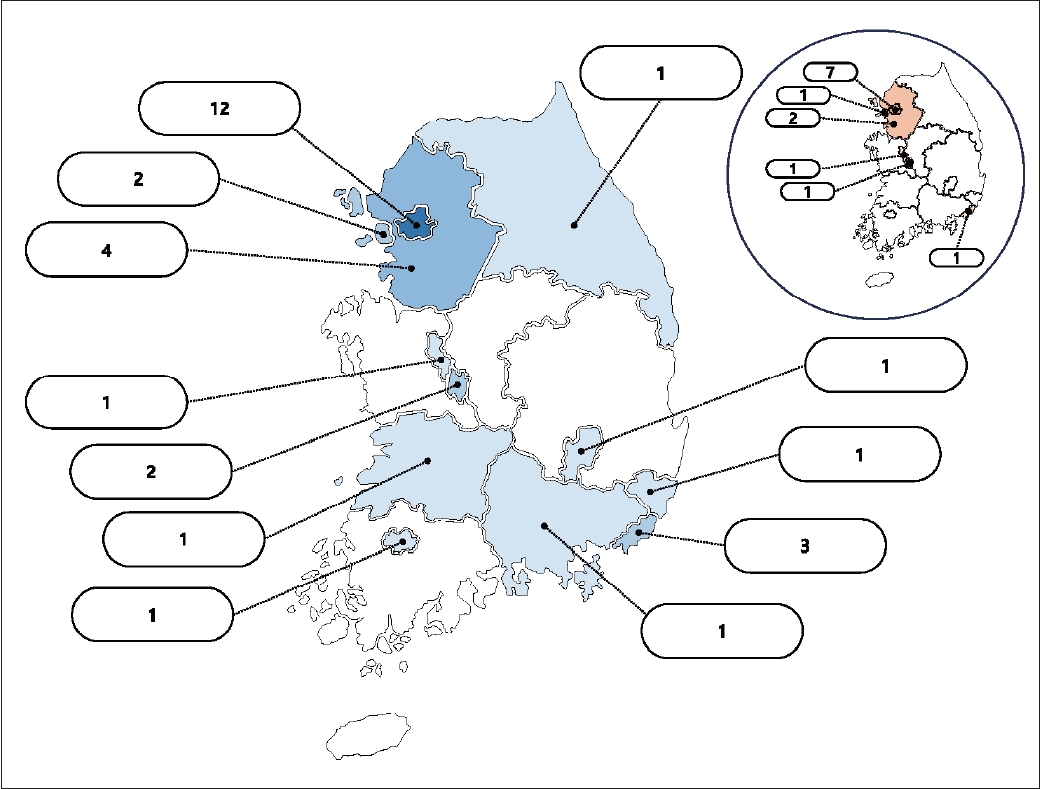
- In an effort to enhance clinical practice and bolster the medical support system for HD in South Korea, the Korean Huntington’s Disease Society (KHDS) was established in July 2022. This development occurred approximately two years after the initiation of the first Korean Huntington’s Disease Cohort (KHDC) study [6]. A total of 13 centers were involved in constructing the initial cohort. However, following the establishment of the KHDS, the KHDC expanded to include 30 sites and continues to actively recruit additional sites for participation (Figure 1).
- This study serves as preliminary groundwork for the development of clinical guidelines for HD in South Korea. Subsequently, the taskforce of the KHDS on the Clinical Management of HD compiled fundamental and updated knowledge concerning the diagnosis and treatment of patients with HD. The initial draft provided by the taskforce underwent meticulous review by a panel of esteemed experts from the Korean Movement Disorders Society. Our aim is for this comprehensive effort to assist clinical practitioners in South Korea in making informed and efficient decisions based on the current resources available in the country. Moreover, we welcome any suggestions and proposals to further refine and advance the clinical guidelines for HD in South Korea in the future.
PREFACE: OVERVIEW OF HUNTINGTON’S DISEASE IN SOUTH KOREA
- Prevalence and incidence of HD
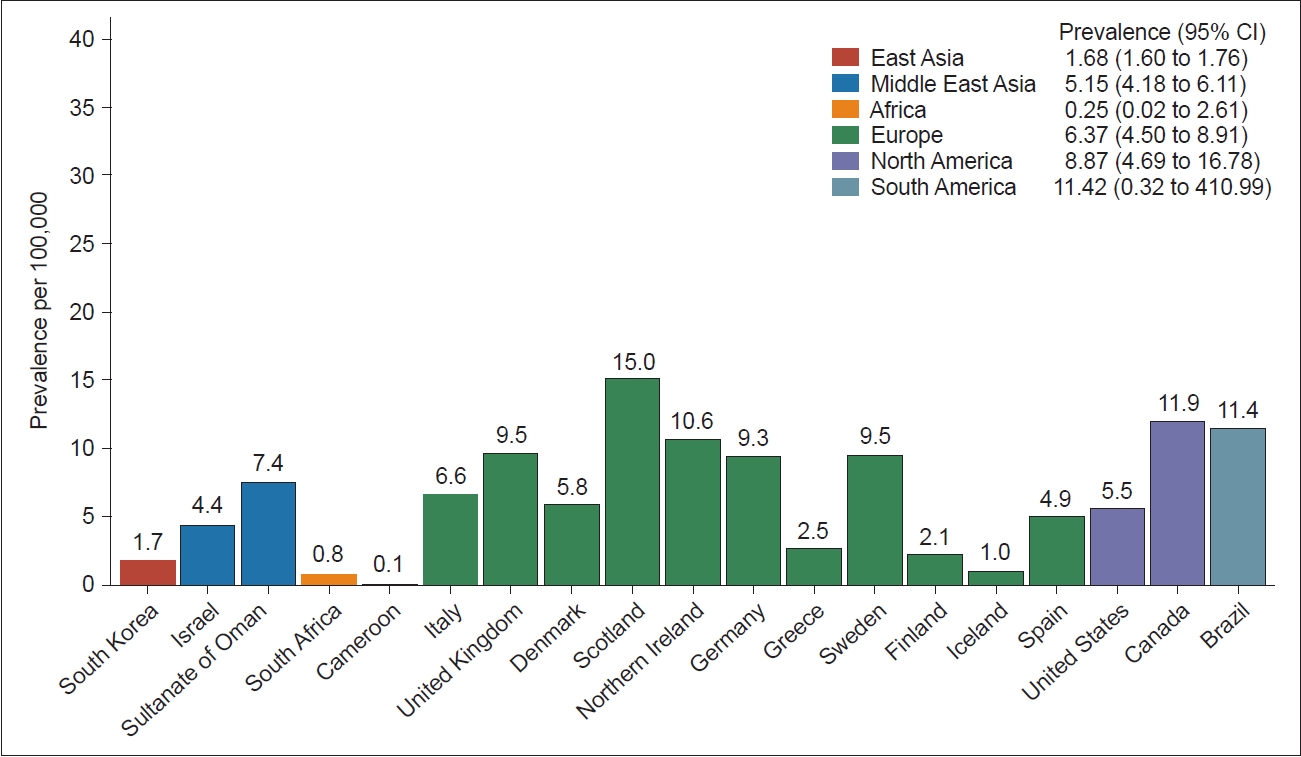
- Over the years, numerous studies have endeavored to estimate the prevalence and incidence of HD across various countries and regions. A meta-analysis encompassing publications from 1985 to 2010 reported a pooled incidence of HD of 0.38 per 100,000 person-years, with a global overall prevalence of 2.71 per 100,000 person-years [7]. In a more recent meta-analysis including studies from 2011 to 2022, there was a slight increase in both indices, with an annual HD incidence of 0.48 per 100,000 person-years and a global overall prevalence of 4.88 per 100,000 person-years [8]. Although these ratios demonstrated a marginal increase compared to previous meta-analyses, the difference was not statistically significant [8]. An improved rate of diagnosis, due to the wider availability of genetic testing, as well as aging populations and increased patient survival rates, might contribute to the observed increase in prevalence and incidence.
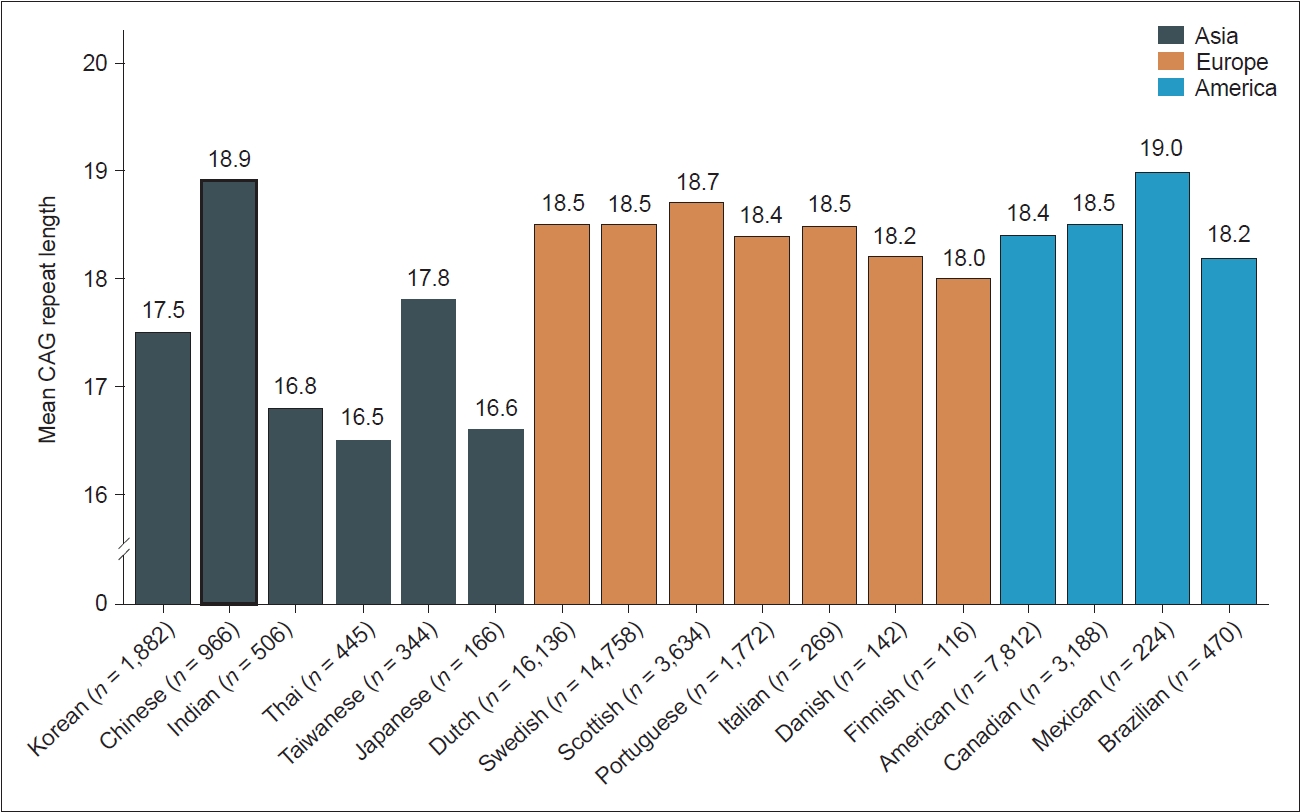
- Significant variations in the prevalence and incidence of HD are observed between countries and regions, suggesting ethnic disparities. Europe and North America exhibit notably higher HD incidence rates than Asia. In addition, Europe, North America, and Oceania had higher prevalence rates than Asia and Africa (Figure 2). These discrepancies suggest that HD occurs less frequently within the Asian population than in Western populations. However, a notable divergence in HD occurrence exists between the Middle East and East Asia; the prevalence and incidence rates in the Middle East closely resemble those in Europe, North America, and Oceania, while rates in East Asia are notably lower (Figure 2) [7,8]. These disparities in HD prevalence across distinct ancestral populations are believed to stem from genetic variations at the HTT locus [9]. Generally, populations with a higher HD prevalence tend to exhibit a greater average length of CAG repeats in the huntingtin (HTT) gene. As illustrated in Figure 3, European populations typically exhibit a range of 18.0–18.7 repeats, whereas most studied Asian populations exhibit a repeat size of lower than 18 [10-21]. In addition to differences in CAG repeat length, differences in the frequencies of haplotypes and CCG polymorphisms in the HTT gene may also be attributed to geographic and ethnic variations in HD epidemiology [22].
- A recent survey in South Korea estimated the prevalence of HD to be approximately 2.22 per 100,000 people, with an annual incidence of 0.29 per 100,000 person-years [5]. These figures indicate lower rates compared to those reported in Western populations but higher rates than previously expected based on earlier reports.
- Frequency of reduced penetrance and intermediate alleles in general populations
- HD patients typically exhibit 36 or more CAG repeats within the HTT gene. However, 36–39 CAG repeats are associated with reduced penetrance, resulting in a later age at onset and slower disease progression than those observed in patients with full penetrance (≥ 40 CAG repeats) [9]. In addition, alleles with between 27 and 35 CAG repeats are classified as intermediate alleles, which are prone to genetic instability and might expand into the disease-causing range within one generation [9]. A comprehensive analysis across three population-based cohorts from British Columbia, the United States, and Scotland revealed that among 7,315 asymptomatic individuals, 0.25% (with an allele frequency of 0.12%) exhibited reduced penetrance, while 6.20% (with an allele frequency of 3.13%) harbored intermediate alleles [11]. This prevalence pattern was corroborated by another study involving five large European population-based cohorts [14], which had similar frequencies of reduced penetrance and intermediate alleles.
- In the South Korean population, among 941 asymptomatic individuals, reduced penetrance and intermediate allele frequencies were notably lower, at 0.11% (with an allele frequency of 0.05%) and 1.38% (with an allele frequency of 0.69%), respectively [10]. This finding mirrors the lower prevalence of HD in South Korea than in Western countries.
- In other Asian populations, intermediate alleles were found in 2.6% of 966 alleles among Chinese individuals [11] and in 0.5% of 430 alleles among Thai individuals [13]. Interestingly, a recent study indicated that East Asian individuals had the lowest prevalence of intermediate alleles (0%, 0/258) among diverse global populations [18]. However, the inclusion of only a limited number of East Asian individuals in these studies suggests the need for a larger sample study to accurately determine the true prevalence in this population.
EPIDEMIOLOGY
- Molecular pathogenesis
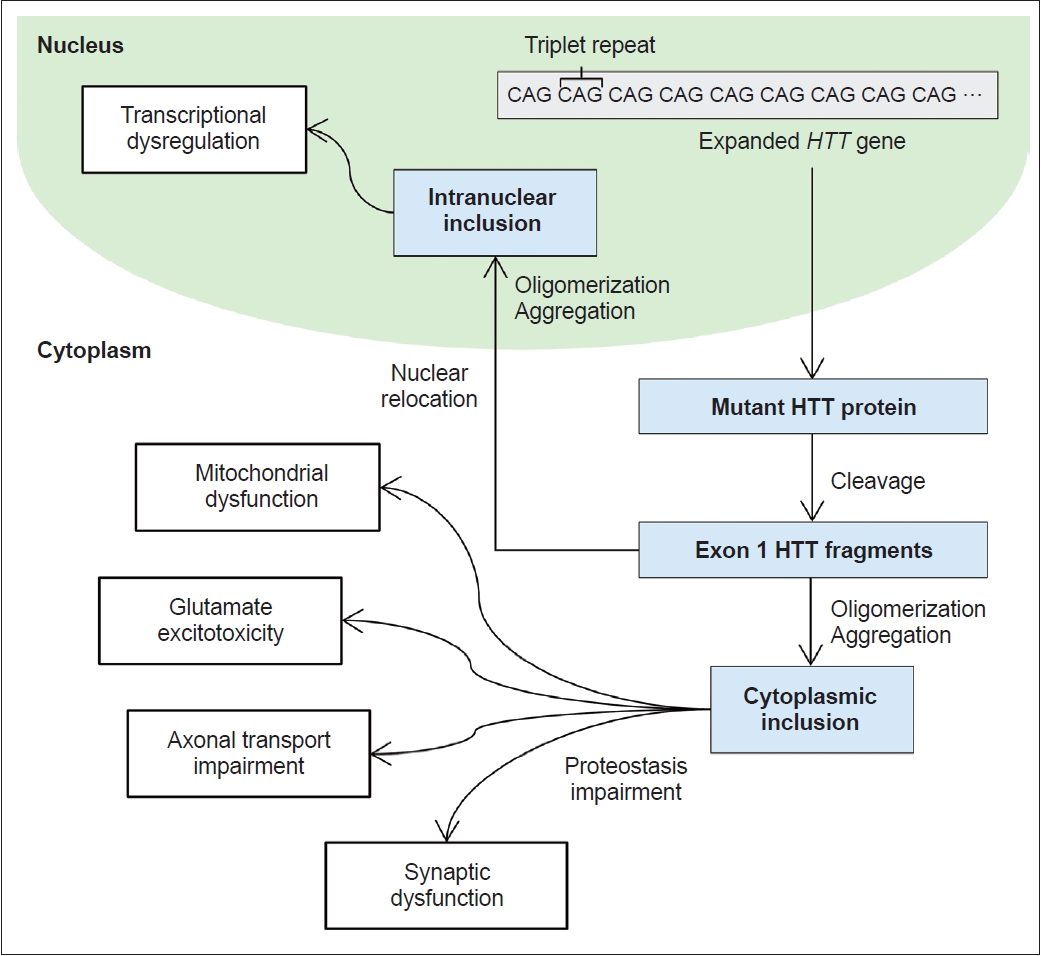
- Extensive data support the pivotal role of mutant HTT (mHTT) fragmentation in the pathogenic mechanism of HD [23]. mHTT fragments originate either from an abnormal splicing event leading to the formation of the HTT exon 1 protein or through the cleavage of full-length HTT by enzymes such as caspases, calpains, and other proteases (Figure 4) [24]. These mHTT fragments instigate neuronal dysfunction and cell death through various pathways, mainly through direct effects from the exon 1 mHTT fragment, the tendency of mHTT to cause abnormal aggregation, and the negative impact of mHTT on cellular proteostasis, axonal transport, transcription and translation, and mitochondrial and synaptic function [25]. Other potential pathogenic mechanisms have been proposed, including reduced levels of brain-derived neurotrophic factor, glutamate excitotoxicity from cortico-striatal projections, and the deleterious effects of repeatassociated non-ATG translation proteins [26,27].
- The differential expression levels of HTT across various cell types contribute to variations in the concentrations of mHTT fragments. Neurons typically exhibit greater HTT expression than glial cells, a factor likely contributing to the predominant neuronal pathology observed in HD [28]. Among the neuronal cells, medium spiny neurons (MSNs) located in the striatum are particularly susceptible to the harmful effects of mHTT [29]. Striatal pathology in HD progresses through two phases: an early phase characterized by the loss of MSNs from the indirect pathway, leading to a hyperkinetic phenotype, and a late phase involving the loss of MSNs from the direct pathway, resulting in a hypokinetic phenotype [30]. The precise mechanism underlying the selective vulnerability of MSNs from the indirect pathway in the early phase is incompletely understood. However, dopamine D2 receptors, which are selectively expressed in indirect MSNs, may substantially contribute to the onset and development of HD [31].
- Macroscopic and microscopic pathology
- Postmortem investigations have revealed a diffuse pattern of atrophy primarily affecting the caudate and putamen in HD patients. This degeneration manifests along gradients on the caudo-rostral, dorso-ventral, and medio-lateral axes. While the globus pallidus and nucleus accumbens are also impacted, the effects are comparatively less pronounced [32]. A classification system has been proposed for the pathologic stages of HD, consisting of the following five grades (0–4) [33]:
- • Grade 0: Clinical signs of HD are present, yet no observable microscopic or macroscopic abnormalities related to the disease are evident.
- • Grade 1: Microscopic observation reveals moderate fibrillary astrocytosis without macroscopic abnormalities in the caudate or putamen.
- • Grade 2: Macroscopic changes become evident in the caudate and putamen but not in the globus pallidus.
- • Grade 3: Fibrillary astrocytosis is observed in the lateral segment of the globus pallidus without involvement of the medial segment.
- • Grade 4: Macroscopic changes include a shrunken and yellow‒ brown caudate, an expanded anterior horn of the lateral ventricle, and a reduced nucleus accumbens. Additional changes may occur in other brain regions, such as the thalamus, subthalamic nucleus, white matter, and cerebellum, particularly in Grades 3 and 4.
- Previous studies employing magnetic resonance imaging have provided supportive evidence for this pathological classification system of HD [34].
PATHOPHYSIOLOGY
- Motor features
- Chorea is the prototypical and most common motor manifestation of HD, occurring in 90% of affected patients [35]. Chorea is more common in adult patients than in juvenile patients, and it typically initiates in the early stages, plateaus, and regresses during the late stages of the disease. In the early stages of HD, chorea is subtle in the extremities, and it is either unnoticed (i.e., anosognosia) independent of cognitive dysfunction, or its presence is denied by the patient, who may camouflage it by semipurposeful movements (i.e., parakinesia). Forehead chorea manifests as enlarged palpebral fissures with eyebrow elevation and frontalis contractions. Motor impersistence is one of the cardinal features of HD and is associated with insuppressible overactivity in HD patients [36]. It leads to an inability to maintain voluntary muscle contraction at a steady level when the patient is asked to maintain tongue protrusion (“flycatcher’s tongue”) or handshake (“milkmaid’s grip”) [36]. Symptoms of chorea in HD patients vary in severity, affecting other motor and nonmotor symptoms (NMSs), daily activities, hospitalization, and quality of life [36,37]. As chorea progresses, patients experience recurrent fall injuries due to postural instability and gait disturbance [38]. Moderate to severe chorea might lead to numerous NMSs, including pain, sleep disturbance, nutritional deficits, and weight loss, as well as social embarrassment and difficulties in communication [35].
- The prevalence of dystonia is reported to be 91%–95% in adult patients, and the most affected body region is the upper limbs, with internal rotation of the shoulder and sustained fist clenching [39,40]. Dystonia worsens as the disease progresses, and its severity is correlated with disease duration and the use of antidopaminergic agents [39].
- Myoclonus is commonly observed in patients with juvenileonset HD [35] and is rarely reported as a predominant and disabling motor feature in patients with adult-onset HD [41]. Myoclonus in HD can be generalized, multifocal, or action-induced cortical myoclonus [42].
- Tics in HD are reported in both juvenile-onset and adult HD patients [43]. However, the pathophysiology of the relationship between HD and Tourette syndrome (TS) needs further investigation to determine whether TS and HD are comorbid or whether TS is an atypical manifestation of HD.
- As the disease progresses, chorea often spontaneously subsides in HD patients. However, parkinsonism can develop and progress to akinesia, severe rigidity, and mutism in the final stages [35]. Since symptomatic treatment for chorea involves reducing dopaminergic transmission by either blocking dopamine receptors or depleting presynaptic dopamine, it may impair motor functions, leading to drug-induced parkinsonism in HD patients [44]. Therefore, hyperkinetic and hypokinetic motor symptoms in HD patients require balanced treatment based on the functional consequences of these movement disorders in individual patients. Late-onset HD patients could be misdiagnosed as having atypical parkinsonism due to their variable motor features, such as dystonia, ataxia, and abnormal oculomotor findings, as well as numerous NMSs, including depression, dementia, and dysautonomia [45].
- In HD, the progressive deterioration of voluntary motor control is prevalent in gait, balance, coordination, oculomotor function, swallowing, and speech. This deterioration is correlated with an acceleration of functional decline [35].
- Cognitive features
- Cognitive dysfunction in HD patients can precede clinical diagnosis by 15 years, and gradual deterioration of cognitive function is highly predictive of typical motor symptom development [46]. According to the REGISTRY study, approximately 8% of patients reported cognitive impairment as the first symptom, while 13% reported a mixed onset of motor, cognitive, and psychiatric symptoms [47]. Cognitive features of HD begin with nonamnestic mild cognitive impairment and progress into a broad range of cognitive deficits in the executive, learning and memory, attention, perception, and language domains [48]. In South Korea, the prevalence of dementia in HD patients is approximately 40% and gradually increases to 80% in patients over 80 years of age [5]. Cognitive performance is poorer in the parkinsonism-dominant group than in the chorea-dominant and mixed-motor phenotype groups, independent of disease duration and severity [49]. Interestingly, patients are often unaware of their cognitive problems. Therefore, physicians should take a history from caregivers, be alert to cognitive behavioral changes and share adaptive strategies with caregivers to maintain patients’ functional capabilities [35].
- Executive dysfunctions encompass slow cognitive processing speed, attentional deficits, and deterioration in decision-making, planning, organization, and sequencing [50].
- Difficulties in the retrieval of knowledge and the acquisition of procedural information are characteristic features [35]. Compared to people with Alzheimer’s dementia (AD), individuals with HD showed better performance on yes/no recognition testing but not on free delayed recall [51]. HD patients exhibit predominant retrieval deficits, whereas AD patients exhibit memory deficits primarily in encoding and storage [50,51]. HD patients can exhibit deficits in implicit memory, which is a collection of coordinated movements and skills, e.g., riding a bicycle, driving a car, chewing, and swallowing [35]. Manifest HD patients have shown impairments in verbal, episodic, visuospatial, prospective, and echoic memory [52].
- Both premanifest and very early manifest HD patients can exhibit an inability to perceive information [35]. This may include recognition of facial emotional expressions and odors, an understanding of time, visuospatial perception, and overall awareness [53].
- HD patients may exhibit delays in initiating speech, decreased syllable rates, reduced numbers of words produced, diminished levels of syntactic complexities, and increased paraphrasing errors with word-finding difficulties [54]. Metabolic imaging revealed that impaired linguistic processing in HD patients was associated with the left striatum and specific portions of the striatum [55].
- Psychiatric features
- Psychiatric features are highly prevalent in both premanifest and manifest HD [35,56]. In adult-onset HD patients, the initial manifestation is more likely to be motor than psychiatric, while juvenile-onset HD patients are equally likely to present with motor, cognitive, or psychiatric features [56]. Psychiatric disturbances in HD patients are often underdiagnosed, leading to inadequate treatment [57] despite their debilitating impacts on patients and their families, potentially causing financial exploitation and hospital admissions [37,58]. Unlike continuously worsening motor and cognitive functions, affective and behavioral disorders show an irregular pattern of deterioration as the disease progresses [56]. Among various psychiatric symptoms, depression, apathy, and irritability are prevalent across all stages of HD, while hallucinations and delusions occur more often in advanced stages of the disease [58].
- Other nonmotor features
- NMSs and signs of HD include a range of metabolic alterations (weight loss), disrupted circadian rhythm (sleep disturbance), and dysautonomia (cardiovascular, gastrointestinal, and genitourinary disorders). Patients with HD experience weight loss, bowel problems, and vivid dreams even more frequently than those with Parkinson’s disease [59]. NMSs in HD can occur before the onset of motor symptoms and correlate with disease duration, total functional capacity, and disease stage. NMSs in HD affect patients’ quality of life with a variable level of importance.
CLINICAL SPECTRUM OF HD
Chorea
Dystonia
Myoclonus
Tourette syndrome
Parkinsonism
Impaired voluntary motor control
Executive function
Learning and memory
Perception
Language
- Clinical diagnosis of HD
- Diagnosis is based on family history, personal history, neurological and psychiatric examinations, and genetic and any other appropriate testing. The diagnostic schemes for presymptomatic, prodromal and manifest HD are summarized in Table 1. Genetic confirmation of HD is based on a CAG expansion of 36 or more repeats in the HTT gene. Full penetrance of HD in mutation carriers is exemplified by > 39 CAG repeats, reduced penetrance is observed between 36–39 repeats, while 35 or fewer repeats are considered normal [26].
- Evaluation of motor dysfunction and severity
- Motor dysfunction and severity are evaluated with the Unified Huntington’s Disease Rating Scale (UHDRS), including total functional capacity, functional assessment, and independence subscales. The UHDRS-Total Motor Score (TMS) assesses eye movements, speech, alternating hand movements, dystonia, chorea and gait. The UHDRS-TMS is sensitive to changes in motor function over time. The details of these scales are explained below [60]. Of note, motor abnormalities in HD patients are rated on a “diagnostic confidence” scale (0–4) according to the probability of manifestation and not according to TMS scores.
- Evaluation of cognitive impairment
- A screening test such as the Mini-Mental State Examination or Montreal Cognitive Assessment (MoCA) may demonstrate only minor changes. The MoCA is perhaps the simplest and most widely used screening test. The Symbol Digit Modalities Test and Stroop Word Reading Test are the best tools for evaluating cognitive function in HD patients. Moreover, the verbal fluency test (category), Stroop color naming test, Stroop interference test, trail making test (Parts A and B), and verbal fluency test (letters) can be used to evaluate cognition in HD patients.
- To diagnose 3 categories of HD (Table 1), the presence of “major cognitive disorder” in individuals needs to be determined based on the DSM-5 criteria as follows. First, modest cognitive decline from a previous level of performance is documented in one or more cognitive domains. Second, cognitive impairment interferes with independence in everyday activities. Comparatively, “minor cognitive disorder” in individuals with HD is defined as cognitive decline that does not interfere with independence but requires greater effort, compensatory strategies, or accommodation. Secondary cognitive impairment due to depression has to be ruled out [61-63].
- Evaluation of neuropsychiatric symptoms
- The Neuropsychiatric Inventory (NPI) assesses the frequency (4-point rating scale) and severity (3-point scale) of 10 neuropsychiatric disturbances (delusions, hallucinations, agitation, dysphoria, anxiety, euphoria, apathy, disinhibition, irritability, and aberrant motor behavior). The NPI offers advantages over previous psychiatric research in HD. Behavior symptoms can be assessed with the problem behavior assessment (short), hospital anxiety and depression scale, Snaith irritable scale and Columbia suicide severity rating scale [63].
- Unified Huntington’s Disease Rating Scale
- The UHDRS was developed by the Huntington Study Group to assess the clinical features of HD patients [64]. The UHDRS is composed of 6 sections (motor, cognitive, behavioral, functional assessment, independence scale, and total functional capacity). A Korean version of the UHDRS is available [6,65].
- The UHDRS-TMS is composed of 15 items and has a maximum score of 124. The items of the UHDRS-TMS include chorea, dystonia, parkinsonism, motor performance, oculomotor function, and balance. The original version was published in 1996 and was updated and expanded in 1999 with the intention of increasing its applicability. Different item combinations of the UHDRS-TMS have been used: 4 shortened versions were published 1 year later (TMS1–4), including a modified motor score as well as reported subitem scores focused on gait, chorea, and dystonia or items related to bradykinesia. The internal consistency of the UHDRS-TMS has been reported to be very good in patients with manifest HD (Cronbach’s alpha 0.95–0.97) [64,66]. The test–retest reliability (0.96 and 0.97) also seems to be very good in patients with manifest HD, although studies have reported correlation coefficients [66]. Interrater reliability in patients with manifest HD is very good, with an intraclass correlation coefficient of 0.94, albeit in a small sample study (n = 24) [64]. In the same study, the interclass correlation coefficient was lower for the chorea (0.82) and dystonia (0.62) subscores [64]. As expected, the UHDRS-TMS score was negatively correlated with the UHDRS-Total Functional Capacity Scale score and disease stage [65], as well as with other UHDRS functional and cognitive scales.
CLINICAL ASSESSMENT OF HD
- Natural course and progression of HD
- After clinical manifestation, HD patients exhibit steady neurological deterioration. Many studies have attempted to measure the progression of HD [67-70]. The annual progression rates derived from the results of these studies are summarized in Table 2. Notably, although most neurological symptoms steadily deteriorate after onset, chorea progresses quickly in the early stages and reaches a plateau before worsening as the disease progresses [70].
- Due to heterogeneity in the progression of diverse clinical symptoms in HD patients, simple functional staging is useful for clinical practice. The Shoulson and Fahn Staging Scale (SF scale) classifies the progression of HD into 5 stages based on the total functional capacity score of the UHDRS (Table 3) [71,72]. However, the SF 5-step classification is not based on meaningful biological deterioration or clinical impact, such as in cancer staging; there is an argument that it is better to use simple 3-step clinical stages (Table 4) than SF staging [73]. Recently, the HD Integrated Staging System, which comprises a biological research definition and evidence-based staging centered on biological, clinical, and functional assessments, was introduced (Figure 5) [74].
- The most valid factors associated with the natural course of HD are age at onset and CAG repeat length. One study reported that younger age at onset is related to faster rates of motor, cognitive, and functional progression [75]. Another study revealed that patients with late-onset HD had a much faster progression rate than patients with usual HD, reaching the severe stage an average of 2.8 years earlier [76]. The effect of CAG repeat length is more complex than that of onset age and has shown controversial results. For example, one study reported that the CAG repeat length was correlated with rapid progression [77], while another study reported the opposite results [78]. In 2019, a longitudinal study in which 443 HD patients were followed up for up to 6 years reported that motor-cognitive function and volumes of the caudate and putamen and the white matter ventricle were associated with CAG repeat length and patient age [79]. Based on the evidence to date, the CAG repeat length is likely to be the most reliable predictor of progression rate in HD patients.
- There are few studies on HD patients in advanced stages. Patients with advanced HD present severe motor and cognitive impairments requiring nursing home placement in most cases. Therefore, neurologists cannot evaluate patients properly at this stage. Caregivers are often unwilling to participate in research because they are exhausted from long-term care for this disastrous disease or become patients of manifest HD. Therefore, further studies on the advanced and terminal stages of patients with HD are needed.
- Survival of HD patients
- The approximate life expectancy of HD patients is known to be approximately 15–20 years. In South Korea, one study analyzed the survival of 47 patients with genetically confirmed HD in 2016 [80]. The mean age at onset was 46.1 ± 14.0 years, and the mean age at death was 57.8 ± 13.7 years. The median survival was 14.5 years. The median survival of HD patients without genetic confirmation has been reported to be 15–18 years in Western populations, and a European HD network cohort study revealed a median survival of 35 years from symptom onset [81]. The shorter survival in South Korea may be explained by the greater mean age at onset, differences in the HTT haplotypes and CCG polymorphisms, influence of other sociocultural factors, and population-specific comorbidities [80].
- In addition to disease progression, age at onset is known to be a predictor of survival in HD patients. Patients with juvenile-onset (younger than 20 years) and late-onset (older than 50 years) disease were reported to have shorter disease durations than HD patients with common onset (20–50 years) [82]. CAG repeat length was found to predict shorter survival in a study reported in 2022 [83]. This study also reported that older age and male sex predicted shorter survival. A recent study in South Korean patients reported that survival after disease onset was shorter in patients with late-onset HD (age at onset ≥ 60 years) than in those with common-onset HD, and longer CAG repeats and greater age at onset were associated with shorter survival in South Korean HD patients [84].
NATURAL COURSE
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This work was supported by a focused clinical research grant-in-aid from the Seoul Metropolitan Government-Seoul National University (SMG-SNU) Boramae Medical Center (04-2022-0014) and a research fund of the Chungnam National University Sejong Hospital (2023-S2-008).
-
Author contributions
Conceptualization: Jee-Young Lee. Data curation: Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Formal analysis: Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Funding acquisition: Jee-Young Lee, Chaewon Shin. Investigation: Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Methodology: Jee-Young Lee, Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Project administration: Jee-Young Lee, Chaewon Shin. Resources: Jee-Young Lee. Supervision: Jee-Young Lee, Jong-Min Kim, Seong-Beom Koh, Manho Kim, Beomseok Jeon. Validation: Jee-Young Lee, Jong-Min Kim, Seong-Beom Koh, Manho Kim, Beomseok Jeon. Visualization: Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Writing—original draft: Jee-Young Lee, Chaewon Shin, Ryul Kim, Dallah Yoo, Eungseok Oh, Jangsup Moon, Minkyeong Kim. Writing—review & editing: Jee-Young Lee, Jong-Min Kim, Seong-Beom Koh, Manho Kim, Beomseok Jeon.
Notes
- None
Acknowledgments

It is expected that the ability to define signs and symptoms would be enhanced by longitudinal follow-up and assessments (modified from Ross et al. Mov Disord Clin Pract 2019;6:541-546. [61], under the terms of the Creative Commons Attribution Non-Commercial License [CC BY NC]). Following the diagnostic confidence level of the Unified Huntington’s Disease Rating Scale: 0, normal (no motor abnormalities); 1, nonspecific motor abnormalities; 2, motor abnormalities that may be signs of HD (50%–89% confidence); 3, motor abnormalities that are likely signs of HD (90%–98% confidence); 4, motor abnormalities that are unequivocal signs of HD (> 99% confidence).
HD, Huntington’s disease.
- 1. Huntington G. On chorea. Med Surg Rep 1872;26:317–321.
- 2. Lee HS, Baek SW, Kim SW. Two cases of probable Huntington’s disease. J Korean Neurol Assoc 1988;6:289–294.
- 3. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993;72:971–983.ArticlePubMed
- 4. Jeon BS, Choi SH, Kim MH, Joo SI, Park SS. Gene analysis in Huntington disease. J Korean Neurol Assoc 1996;14:494–501.
- 5. Lee CY, Ro JS, Jung H, Kim M, Jeon B, Lee JY. Increased 10-year prevalence of Huntington’s disease in South Korea: an analysis of medical expenditure through the national healthcare system. J Clin Neurol 2023;19:147–155.ArticlePubMedPMCPDF
- 6. Lee CY, Shin C, Hwang YS, Oh E, Kim M, Kim HS, et al. Caregiver burden of patients with Huntington’s disease in South Korea. J Mov Disord 2024;17:30–37.ArticlePubMedPDF
- 7. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 2012;27:1083–1091.ArticlePubMed
- 8. Medina A, Mahjoub Y, Shaver L, Pringsheim T. Prevalence and incidence of Huntington’s disease: an updated systematic review and meta-analysis. Mov Disord 2022;37:2327–2335.ArticlePubMedPMCPDF
- 9. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers 2015;1:15005.ArticlePubMedPDF
- 10. Kim R, Seong MW, Oh B, Shin HS, Lee JS, Park S, et al. Analysis of HTT CAG repeat expansion among healthy individuals and patients with chorea in Korea. Parkinsonism Relat Disord 2024;118:105930.ArticlePubMed
- 11. Jiang H, Sun YM, Hao Y, Yan YP, Chen K, Xin SH, et al. Huntingtin gene CAG repeat numbers in Chinese patients with Huntington’s disease and controls. Eur J Neurol 2014;21:637–642.PubMed
- 12. Pramanik S, Basu P, Gangopadhaya PK, Sinha KK, Jha DK, Sinha S, et al. Analysis of CAG and CCG repeats in Huntingtin gene among HD patients and normal populations of India. Eur J Hum Genet 2000;8:678–682.ArticlePubMedPDF
- 13. Pulkes T, Papsing C, Wattanapokayakit S, Mahasirimongkol S. CAG-expansion haplotype analysis in a population with a low prevalence of Huntington’s disease. J Clin Neurol 2014;10:32–36.ArticlePubMedPMC
- 14. Wang CK, Wu YR, Hwu WL, Chen CM, Ro LS, Chen ST, et al. DNA haplotype analysis of CAG repeat in Taiwanese Huntington’s disease patients. Eur Neurol 2004;52:96–100.ArticlePubMedPDF
- 15. Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet 1994;3:2103–2114.ArticlePubMed
- 16. Sundblom J, Niemelä V, Ghazarian M, Strand AS, Bergdahl IA, Jansson JH, et al. High frequency of intermediary alleles in the HTT gene in Northern Sweden - the Swedish Huntingtin Alleles and Phenotype (SHAPE) study. Sci Rep 2020;10:9853.ArticlePubMedPMCPDF
- 17. Gardiner SL, Boogaard MW, Trompet S, de Mutsert R, Rosendaal FR, Gussekloo J, et al. Prevalence of carriers of intermediate and pathological polyglutamine disease–associated alleles among large population-based cohorts. JAMA Neurol 2019;76:650–656.ArticlePubMed
- 18. Kay C, Collins JA, Wright GEB, Baine F, Miedzybrodzka Z, Aminkeng F, et al. The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population. Am J Med Genet B Neuropsychiatr Genet 2018;177:346–357.ArticlePubMedPDF
- 19. Costa MDC, Magalhães P, Guimarães L, Maciel P, Sequeiros J, Sousa A. The CAG repeat at the Huntington disease gene in the Portuguese population: insights into its dynamics and to the origin of the mutation. J Hum Genet 2006;51:189–195.ArticlePubMedPDF
- 20. Kay C, Collins JA, Miedzybrodzka Z, Madore SJ, Gordon ES, Gerry N, et al. Huntington disease reduced penetrance alleles occur at high frequency in the general population. Neurology 2016;87:282–288.ArticlePubMedPMC
- 21. Alonso ME, Ochoa A, Boll MC, Sosa AL, Yescas P, López M, et al. Clinical and genetic characteristics of Mexican Huntington’s disease patients. Mov Disord 2009;24:2012–2015.ArticlePubMedPDF
- 22. Xu M, Wu ZY. Huntington disease in Asia. Chin Med J (Engl) 2015;128:1815–1819.ArticlePubMedPMC
- 23. Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol 2020;16:529–546.ArticlePubMedPDF
- 24. Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98.ArticlePubMed
- 25. Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med 2017;7:a024240.ArticlePubMedPMC
- 26. McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol 2018;25:24–34.ArticlePubMedPDF
- 27. Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, et al. RAN translation in Huntington disease. Neuron 2015;88:667–677.ArticlePubMedPMC
- 28. Wilton DK, Stevens B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol Dis 2020;143:104963.ArticlePubMedPMC
- 29. Morigaki R, Goto S. Striatal vulnerability in Huntington’s disease: neuroprotection versus neurotoxicity. Brain Sci 2017;7:63.ArticlePubMedPMC
- 30. Plotkin JL, Surmeier DJ. Corticostriatal synaptic adaptations in Huntington’s disease. Curr Opin Neurobiol 2015;33:53–62.ArticlePubMedPMC
- 31. Deyts C, Galan-Rodriguez B, Martin E, Bouveyron N, Roze E, Charvin D, et al. Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtininduced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS One 2009;4:e8287.ArticlePubMedPMC
- 32. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol 1998;57:369–384.ArticlePubMed
- 33. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985;44:559–577.ArticlePubMed
- 34. Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10:31–42.ArticlePubMed
- 35. Nance MA, Paulsen JS, Rosenblatt A, Wheelock V. A physician’s guide to the management of Huntington’s disease. 3rd ed. New York: Huntington’s Disease Society of America; 2011.
- 36. Stoker TB, Mason SL, Greenland JC, Holden ST, Santini H, Barker RA. Huntington’s disease: diagnosis and management. Pract Neurol 2022;22:32–41.ArticlePubMed
- 37. Peball M, Heim B, Ellmerer P, Frank F, Busin N, Galffy M, et al. Hospital admissions of Huntington’s disease patients in a Huntington’s disease centre between 2011 and 2016: a retrospective analysis. Mov Disord Clin Pract 2022;9:628–636.ArticlePubMedPMCPDF
- 38. Kloos AD, Kegelmeyer DA, Young GS, Kostyk SK. Fall risk assessment using the Tinetti mobility test in individuals with Huntington’s disease. Mov Disord 2010;25:2838–2844.ArticlePubMedPDF
- 39. Louis ED, Lee P, Quinn L, Marder K. Dystonia in Huntington’s disease: prevalence and clinical characteristics. Mov Disord 1999;14:95–101.ArticlePubMed
- 40. van de Zande NA, Massey TH, McLauchlan D, Pryce Roberts A, Zutt R, Wardle M, et al. Clinical characterization of dystonia in adult patients with Huntington’s disease. Eur J Neurol 2017;24:1140–1147.ArticlePubMedPDF
- 41. Thompson PD, Bhatia KP, Brown P, Davis MB, Pires M, Quinn NP, et al. Cortical myoclonus in Huntington’s disease. Mov Disord 1994;9:633–641.ArticlePubMed
- 42. Rossi Sebastiano D, Soliveri P, Panzica F, Moroni I, Gellera C, Gilioli I, et al. Cortical myoclonus in childhood and juvenile onset Huntington’s disease. Parkinsonism Relat Disord 2012;18:794–797.ArticlePubMed
- 43. Müller J, Wenning GK, Wissel J, Poewe W. Intrafamilial heterogeneity of facial hyperkinesias: chance association of tics, cranial dystonia, and Huntington’s disease? Mov Disord 2001;16:370–372.ArticlePubMed
- 44. Reilmann R. Parkinsonism in Huntington’s disease. Int Rev Neurobiol 2019;149:299–306.ArticlePubMed
- 45. Reuter I, Hu MT, Andrews TC, Brooks DJ, Clough C, Chaudhuri KR. Late onset levodopa responsive Huntington’s disease with minimal chorea masquerading as Parkinson plus syndrome. J Neurol Neurosurg Psychiatry 2000;68:238–241.ArticlePubMedPMC
- 46. Paulsen JS, Long JD, Ross CA, Harrington DL, Erwin CJ, Williams JK, et al. Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. Lancet Neurol 2014;13:1193–1201.ArticlePubMedPMC
- 47. Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, et al. Observing Huntington’s disease: the European Huntington’s disease network’s REGISTRY. PLoS Curr 2010;2:RRN1184.ArticlePubMed
- 48. Cavallo M, Sergi A, Pagani M. Cognitive and social cognition deficits in Huntington’s disease differ between the prodromal and the manifest stages of the condition: a scoping review of recent evidence. Br J Clin Psychol 2022;61:214–241.ArticlePDF
- 49. Hart EP, Marinus J, Burgunder JM, Bentivoglio AR, Craufurd D, Reilmann R, et al. Better global and cognitive functioning in choreatic versus hypokinetic-rigid Huntington’s disease. Mov Disord 2013;28:1142–1145.ArticlePubMedPDF
- 50. Snowden JS. The neuropsychology of Huntington’s disease. Arch Clin Neuropsychol 2017;32:876–887.ArticlePubMed
- 51. Fine EM, Delis DC, Wetter SR, Jacobson MW, Hamilton JM, Peavy G, et al. Identifying the “source” of recognition memory deficits in patients with Huntington’s disease or Alzheimer’s disease: evidence from the CVLT-II. J Clin Exp Neuropsychol 2008;30:463–470.ArticlePubMedPMC
- 52. Beste C, Saft C, Güntürkün O, Falkenstein M. Increased cognitive functioning in symptomatic Huntington’s disease as revealed by behavioral and event-related potential indices of auditory sensory memory and attention. J Neurosci 2008;28:11695–11702.ArticlePubMedPMC
- 53. Dumas EM, van den Bogaard SJ, Middelkoop HA, Roos RA. A review of cognition in Huntington’s disease. Front Biosci (Schol Ed) 2013;5:1–18.ArticlePubMed
- 54. Wallesch CW, Fehrenbach RA. On the neurolinguistic nature of language abnormalities in Huntington’s disease. J Neurol Neurosurg Psychiatry 1988;51:367–373.ArticlePubMedPMC
- 55. Teichmann M, Gaura V, Démonet JF, Supiot F, Delliaux M, Verny C, et al. Language processing within the striatum: evidence from a PET correlation study in Huntington’s disease. Brain 2008;131(Pt 4):1046–1056.ArticlePubMed
- 56. McAllister B, Gusella JF, Landwehrmeyer GB, Lee JM, MacDonald ME, Orth M, et al. Timing and impact of psychiatric, cognitive, and motor abnormalities in Huntington disease. Neurology 2021;96:e2395–e2406.ArticlePubMedPMC
- 57. Eddy CM, Parkinson EG, Rickards HE. Changes in mental state and behaviour in Huntington’s disease. Lancet Psychiatry 2016;3:1079–1086.ArticlePubMed
- 58. van Duijn E, Craufurd D, Hubers AA, Giltay EJ, Bonelli R, Rickards H, et al. Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry 2014;85:1411–1418.ArticlePubMed
- 59. Aldaz T, Nigro P, Sánchez-Gómez A, Painous C, Planellas L, Santacruz P, et al. Non-motor symptoms in Huntington’s disease: a comparative study with Parkinson’s disease. J Neurol 2019;266:1340–1350.ArticlePubMedPDF
- 60. Mestre TA, Forjaz MJ, Mahlknecht P, Cardoso F, Ferreira JJ, Reilmann R, et al. Rating scales for motor symptoms and signs in Huntington’s disease: critique and recommendations. Mov Disord Clin Pract 2018;5:111–117.ArticlePubMedPMCPDF
- 61. Ross CA, Reilmann R, Cardoso F, McCusker EA, Testa CM, Stout JC, et al. Movement Disorder Society task force viewpoint: Huntington’s disease diagnostic categories. Mov Disord Clin Pract 2019;6:541–546.ArticlePubMedPMCPDF
- 62. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington’s disease based on natural history. Mov Disord 2014;29:1335–1341.ArticlePubMed
- 63. Anderson KE, van Duijn E, Craufurd D, Drazinic C, Edmondson M, Goodman N, et al. Clinical management of neuropsychiatric symptoms of Huntington disease: expert-based consensus guidelines on agitation, anxiety, apathy, psychosis and sleep disorders. J Huntingtons Dis 2018;7:355–366.ArticlePubMedPMC
- 64. Huntington Study Group. Unified Huntington’s disease rating scale: reliability and consistency. Mov Disord 1996;11:136–142.ArticlePubMed
- 65. Hwang YS, Oh E, Kim M, Lee CY, Kim HS, Chung SJ, et al. Plasma neurofilament light-chain and phosphorylated tau as biomarkers of disease severity in Huntington’s disease: Korean cohort data. J Neurol Sci 2023;452:120744.ArticlePubMed
- 66. Siesling S, Zwinderman AH, van Vugt JP, Kieburtz K, Roos RA. A shortened version of the motor section of the unified Huntington’s disease rating scale. Mov Disord 1997;12:229–234.ArticlePubMed
- 67. Shoulson I, Odoroff C, Oakes D, Behr J, Goldblatt D, Caine E, et al. A controlled clinical trial of baclofen as protective therapy in early Huntington’s disease. Ann Neurol 1989;25:252–259.ArticlePubMed
- 68. Meyer C, Landwehrmeyer B, Schwenke C, Doble A, Orth M, Ludolph AC. Rate of change in early Huntington’s disease: a clinicometric analysis. Mov Disord 2012;27:118–124.ArticlePubMedPDF
- 69. Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 2013;12:637–649.ArticlePubMed
- 70. Dorsey ER, Beck CA, Darwin K, Nichols P, Brocht AF, Biglan KM, et al. Natural history of Huntington disease. JAMA Neurol 2013;70:1520–1530.ArticlePubMed
- 71. Shoulson I. Huntington disease: functional capacities in patients treated with neuroleptic and antidepressant drugs. Neurology 1981;31:1333–1335.ArticlePubMed
- 72. Shoulson I, Fahn S. Huntington disease: clinical care and evaluation. Neurology 1979;29:1–3.ArticlePubMed
- 73. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10:204–216.ArticlePubMedPDF
- 74. Tabrizi SJ, Schobel S, Gantman EC, Mansbach A, Borowsky B, Konstantinova P, et al. A biological classification of Huntington’s disease: the integrated staging system. Lancet Neurol 2022;21:632–644.ArticlePubMed
- 75. Mahant N, McCusker EA, Byth K, Graham S. Huntington’s disease: clinical correlates of disability and progression. Neurology 2003;61:1085–1092.ArticlePubMed
- 76. Koutsis G, Karadima G, Kladi A, Panas M. Late-onset Huntington’s disease: diagnostic and prognostic considerations. Parkinsonism Relat Disord 2014;20:726–730.ArticlePubMed
- 77. Rosenblatt A, Kumar BV, Mo A, Welsh CS, Margolis RL, Ross CA. Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov Disord 2012;27:272–276.ArticlePubMed
- 78. Squitieri F, Cannella M, Simonelli M. CAG mutation effect on rate of progression in Huntington’s disease. Neurol Sci 2002;23(Suppl 2):S107–S108.ArticlePubMedPDF
- 79. Langbehn DR, Stout JC, Gregory S, Mills JA, Durr A, Leavitt BR, et al. Association of CAG repeats with long-term progression in Huntington disease. JAMA Neurol 2019;76:1375–1385.ArticlePubMedPMC
- 80. Kim HJ, Shin CW, Jeon B, Park H. Survival of Korean Huntington’s disease patients. J Mov Disord 2016;9:166–170.ArticlePubMedPMCPDF
- 81. Rodrigues FB, Abreu D, Damásio J, Goncalves N, Correia-Guedes L, Coelho M, et al. Survival, mortality, causes and places of death in a European Huntington’s disease prospective cohort. Mov Disord Clin Pract 2017;4:737–742.ArticlePubMedPMCPDF
- 82. Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry 1999;66:52–56.ArticlePubMedPMC
- 83. Langbehn DR. Longer CAG repeat length is associated with shorter survival after disease onset in Huntington disease. Am J Hum Genet 2022;109:172–179.ArticlePubMed
- 84. Hwang YS, Jo S, Kim GH, Lee JY, Ryu HS, Oh E, et al. Clinical and genetic characteristics associated with survival outcome in late-onset Huntington’s disease in South Korea. J Clin Neurol 2024 Apr 2 [Epub]. Available from: https://doi.org/10.3988/jcn.2023.0329. Article
- 85. Ravina B, Romer M, Constantinescu R, Biglan K, Brocht A, Kieburtz K, et al. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov Disord 2008;23:1223–1227.ArticlePubMedPDF
- 86. Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K, et al. Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology 2000;54:452–458.ArticlePubMed
- 87. Siesling S, van Vugt JP, Zwinderman KA, Kieburtz K, Roos RA. Unified Huntington’s disease rating scale: a follow up. Mov Disord 1998;13:915–919.ArticlePubMedPDF
- 88. Feigin A, Kieburtz K, Bordwell K, Como P, Steinberg K, Sotack J, et al. Functional decline in Huntington’s disease. Mov Disord 1995;10:211–214.ArticlePubMed
- 89. Bamford KA, Caine ED, Kido DK, Cox C, Shoulson I. A prospective evaluation of cognitive decline in early Huntington’s disease: functional and radiographic correlates. Neurology 1995;45:1867–1873.ArticlePubMed
- 90. Penney JB Jr, Young AB, Shoulson I, Starosta-Rubenstein S, Snodgrass SR, Sanchez-Ramos J, et al. Huntington’s disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov Disord 1990;5:93–99.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Comments on this article
JEE YOUNG LEE
March 26, 2024
 PubReader
PubReader ePub Link
ePub Link Cite
Cite





Manho Kim
March 31, 2024