E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(3); 2023 > Article
-
Review Article
Nine Hereditary Movement Disorders First Described in Asia: Their History and Evolution -
Priya Jagota1

 , Yoshikazu Ugawa2, Zakiyah Aldaajani3, Norlinah Mohamed Ibrahim4, Hiroyuki Ishiura5,6, Yoshiko Nomura7, Shoji Tsuji8, Cid Diesta9, Nobutaka Hattori10, Osamu Onodera11, Saeed Bohlega12, Amir Al-Din13, Shen-Yang Lim14,15, Jee-Young Lee16, Beomseok Jeon17,18, Pramod Kumar Pal19, Huifang Shang20, Shinsuke Fujioka21, Prashanth Lingappa Kukkle22,23, Onanong Phokaewvarangkul1, Chin-Hsien Lin24, Cholpon Shambetova25, Roongroj Bhidayasiri1,26
, Yoshikazu Ugawa2, Zakiyah Aldaajani3, Norlinah Mohamed Ibrahim4, Hiroyuki Ishiura5,6, Yoshiko Nomura7, Shoji Tsuji8, Cid Diesta9, Nobutaka Hattori10, Osamu Onodera11, Saeed Bohlega12, Amir Al-Din13, Shen-Yang Lim14,15, Jee-Young Lee16, Beomseok Jeon17,18, Pramod Kumar Pal19, Huifang Shang20, Shinsuke Fujioka21, Prashanth Lingappa Kukkle22,23, Onanong Phokaewvarangkul1, Chin-Hsien Lin24, Cholpon Shambetova25, Roongroj Bhidayasiri1,26 -
Journal of Movement Disorders 2023;16(3):231-247.
DOI: https://doi.org/10.14802/jmd.23065
Published online: June 13, 2023
1Chulalongkorn Centre of Excellence for Parkinson’s Disease and Related Disorders, Department of Medicine, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
2Department of Human Neurophysiology, Faculty of Medicine, Fukushima Medical University, Fukushima, Japan
3Neurology Unit, King Fahad Military Medical Complex, Dhahran, Saudi Arabia
4Neurology Unit, Department of Medicine, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
5Department of Neurology, Faculty of Medicine, The University of Tokyo, Tokyo, Japan
6Department of Neurology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan
7Yoshiko Nomura Neurological Clinic for Children, Tokyo, Japan
8Institute of Medical Genomics, International University of Health and Welfare, Narita, Chiba, Japan
9Section of Neurology, Department of Neuroscience, Makati Medical Center, NCR, Makati City, Philippines
10Department of Neurology, Juntendo University School of Medicine, Tokyo, Japan
11Department of Neurology, Brain Research Institute, Niigata University, Niigata, Japan
12Department of Neurosciences, King Faisal Specialist Hospital & Research Center, Riyad, Saudi Arabia
13Mid Yorkshire Hospitals National Health Services Trust, Wakefield, UK
14Division of Neurology, Department of Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
15The Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
16Department of Neurology, Seoul Metropolitan Government-Seoul National University Boramae Medical Center & Seoul National University Medical College, Seoul, Korea
17Department of Neurology, Seoul National University, Seoul, Korea
18Movement Disorder Center, Seoul National University Hospital, Seoul, Korea
19Department of Neurology, National Institute of Mental Health & Neurosciences (NIMHANS), Bengaluru, Karnataka, India
20Department of Neurology, Laboratory of Neurodegenerative Disorders, Rare Diseases Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
21Department of Neurology, Fukuoka University, Faculty of Medicine, Fukuoka, Japan
22Center for Parkinson’s Disease and Movement Disorders, Manipal Hospital, Bangalore, India
23Parkinson's Disease and Movement Disorders Clinic, Bangalore, India
24Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan
25I. K. Akhunbaev Kyrgyz State Medical Academy, Bishkek, Kyrgyzstan
26The Academy of Science, The Royal Society of Thailand, Bangkok, Thailand
- Corresponding author: Priya Jagota, MD, MSc Chulalongkorn Centre of Excellence for Parkinson’s Disease and Related Disorders, 7th Floor, Sor Tor Building, King Chulalongkorn Memorial Hospital, 1873 Rama 4 Road, Pathum Wan, Bangkok 10330, Thailand/ Tel: +66-2-256-4000 ext. 70701 / Fax: +66-2-256-4630 / E-mail: pja@chulapd.org
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 2,939 Views
- 232 Download
ABSTRACT
- Clinical case studies and reporting are important to the discovery of new disorders and the advancement of medical sciences. Both clinicians and basic scientists play equally important roles leading to treatment discoveries for both cures and symptoms. In the field of movement disorders, exceptional observation of patients from clinicians is imperative, not just for phenomenology but also for the variable occurrences of these disorders, along with other signs and symptoms, throughout the day and the disease course. The Movement Disorders in Asia Task Force (TF) was formed to help enhance and promote collaboration and research on movement disorders within the region. As a start, the TF has reviewed the original studies of the movement disorders that were preliminarily described in the region. These include nine disorders that were first described in Asia: Segawa disease, PARK-Parkin, X-linked dystonia-parkinsonism, dentatorubral-pallidoluysian atrophy, Woodhouse-Sakati syndrome, benign adult familial myoclonic epilepsy, Kufor-Rakeb disease, tremulous dystonia associated with mutation of the calmodulin-binding transcription activator 2 gene, and paroxysmal kinesigenic dyskinesia. We hope that the information provided will honor the original researchers and help us learn and understand how earlier neurologists and basic scientists together discovered new disorders and made advances in the field, which impact us all to this day.
- There is a rich history of movement disorder case reporting in medical science tracing back many generations. The outstanding example is, of course, by James Parkinson, who described six illustrative cases of paralysis agitans, the disorder that now bears his name [1]. Original case reports, reflecting the work of clinicians, provide the foundation upon which medical knowledge is built and have, throughout the centuries, contributed enormously to our understanding of many disease entities. In addition, evidence accumulated from case reports often inspires larger controlled studies that further contribute to advancements in diagnosis and treatment. A good example of this is the original observation of inspiratory sigh and stridor by Professor Niall Quinn in patients with multiple system atrophy (MSA). This observation led to further clinicopathological examinations and eventually a large European MSA study. Later, the observation was incorporated as a feature supporting MSA diagnosis [2-8]. Even though the format of case reports has evolved, the essential elements remain the patient and their story, with associated assessments, followed by an informed discussion that forms a vital part of continuous life-long medical education.
- The Movement Disorders in Asia Task Force (TF) of the International Parkinson Disease and Movement Disorder Society-Asian Oceanian Section (MDS-AOS) was formed in June 2020 to promote and enhance research and collaboration among movement disorder clinicians and basic scientists in the region. The TF has conducted a review of various aspects of movement disorders in the region to provide a basic understanding of the overall conditions encountered in Asia. Here, we re-evaluate the original reports of movement disorder conditions that were first described in Asia. The TF worked with original contributors, or their successors when possible, to provide early descriptions of their cases and reflect on advancements made in these conditions following the original observations. The aim was to highlight the educational impact of these original cases on current clinical and research practice and to contemplate how the use of the key clinical skills demonstrated by those involved in listening, observation, perseverance, curiosity, and sound reasoning has led to the discovery of a new disorder.
INTRODUCTION
- The TF assembled and derived a list of disorders known to be first described in Asia, mainly based on the expert opinion of the TF members. TF members consist of movement disorder specialists from major regions in Asia, including Japan, China, Taiwan, South Korea, India, Thailand, Malaysia, the Philippines, Kyrgyzstan and Saudi Arabia. The original case contributors were contacted, where possible, and were asked to review the conditions they originally reported or to comment on the review written by other authors in the TF. If the original contributors could not be contacted, where possible, we contacted other contributors who were associated with the early stages of the recognition of the disorder.
METHODS
- Nine movement disorder conditions were included: Segawa disease, PARK-Parkin, X-linked dystonia parkinsonism (XDP), dentatorubral-pallidoluysian atrophy (DRPLA), Woodhouse-Sakati syndrome (WSS), benign adult familial myoclonic epilepsy (BAFME), Kufor-Rakeb disease, tremulous dystonia associated with mutation of the calmodulin-binding transcription activator 2 (CAMTA2) gene, and paroxysmal kinesigenic dyskinesia (PKD) (Table 1).
- Segawa disease; DYT/PARK-GCH1; DYT5a
- In 1970, a girl suffering from a walking difficulty that worsened toward the evening visited Dr. Segawa. A female cousin had the same symptoms. Dr. Segawa decided to try levodopa treatment—a drug that had been recently discovered at that time. In 1971, these two girls were reported in a Japanese journal as suffering from “hereditary basal ganglia disease with marked diurnal fluctuation” [9]. When seeing a 51-year-old patient who was the grandmother of one proband with a clinical course of 43 years, Dr. Segawa noticed that their symptom was dystonia, not parkinsonism. Therefore, in 1975, Dr. Segawa reported a revised nomenclature of “hereditary progressive dystonia with marked diurnal fluctuation (HPD)” at the First International Dystonia Congress in New York [10]. However, the disorder became known as “Segawa disease”.
- Dr. Segawa classified the disease into two types: the postural and action types [11]. Patients with the postural type show postural dystonia as the primary symptom throughout the disease course. The onset is at approximately 6 years of age, and the initial symptom is mostly unilateral pes equinovarus. With age, dystonia expands to other limbs and trunk muscles. Patients with the action type develop dystonic movement of one extremity or the neck (action retrocollis) in addition to a dystonic posture at approximately 8 to 10 years of age.
- The marked and sustained response to levodopa without side effects suggested that this disease is a functional disorder of nigrostriatal (NS)-dopamine (DA) neurons [10,12-16]. McGeer and McGeer have shown that tyrosine hydroxylase (TH) activity in the caudate nucleus decreases in an age-dependent manner [17], giving a hint to the pathogenesis of the disorder [12-16]. This suggests that TH is decreased at the terminal of NS-DA neurons and follows the normal decrement [12-16]. The diurnal fluctuation of symptoms may be explained by the circadian oscillation of NS-DA neurons at the terminal (striatum) [17] but not at the perikaryon (substantia nigra) [18]. Sleep studies have confirmed a decrease in DA activity in the striatum [10,12-16,19-21].
- Dr. Segawa proposed [19-22] that the involvement of the direct striatal pathway (via D1 DA receptors) and the subsequent descending pathway from the basal ganglia to the midbrain and brain stem results in dystonia. In contrast, the action type results from the hypofunction of the nigro-subthalamic DA neurons [19-22] that innervate the subthalamic nucleus (STN) via D1 DA receptors in the STN [23]. The net disinhibition of the thalamo-cortical pathway results in dystonia [19-22]. Neuropathological study findings have confirmed the proposed pathophysiology; TH activity is low in the striatum but not in the substantia nigra [22]. Neuropathological and neurohistochemical studies have revealed TH activity and immunoreactive protein reduction in the striatum but not in the substantia nigra [22,24], and the subregional dorsoventral gradient of the DA decrement shows a marked decrease in the ventral area in contrast to the dorsal predominance in PD [25]. This finding suggests that NS-DA neurons connecting to the D1-direct pathways are predominantly affected in Segawa disease [14-16,19-22].
- In 1990, Fujita and Shintaku reported a decrease in biopterin and neopterin in cerebrospinal fluid and suggested a deficiency of guanosine-triphosphate-cyclohydrolase 1 (GCH1) as the cause of Segawa disease [26]. In addition, Furukawa et al. [27] analyzed the CSF of patients with Segawa disease, PD, and other disorders and showed a decrease in biopterin and neopterin only in patients with Segawa disease. Finally, in 1994, Dr. Ichinose discovered the GTP cyclohydrolase1 gene to be the causative gene of Segawa disease [28]. Since then, a few other gene mutations have been identified that cause similar symptoms that are responsive to low-dose levodopa, referred to as dopa-responsive dystonias [29]. Parkinson’s disease symptoms can also be seen in some GTP cyclohydrolase1 gene mutation carriers, suggesting that GCH1 deficiency may predispose patients to NS cell loss [30].
- For treatment, levodopa shows marked and sustained effects without any side effects (Supplementary Video 1 in the online-only Data Supplement) [12-16], but the effect of levodopa may fluctuate in some patients with the action type [11]. Anticholinergics were used before the discovery of levodopa [31], which showed beneficial effects on dystonia but not on tremors. Tetrahydrobiopterin (BH4) may be used for patients who are compound heterozygotes with early-onset cases [32].
- PARK-Parkin; PARK2
- Nasu et al. [33] first reported, based on only clinical features, the cases of four patients with familial juvenile Parkinson’s disease in 1958, with two subsequent reports quickly following this [34,35]. Then, in 1973, based on these Japanese case reports, Yamamura et al. [36] summarized the clinical features of the disorder as one genetic entity when he described the cases of a familial group of patients with early-onset parkinsonism marked by sleep benefit that subsided as the disease progressed. He coined the disorder “paralysis agitans of early onset with marked diurnal fluctuation of symptoms” [36]. Clinical progression was slow, and all patients responded to trihexyphenidyl treatment. One patient treated with levodopa showed a dramatic improvement in her symptoms.
- The discovery of the parkin gene as the responsible genetic link followed the report of a family with young-onset parkinsonism, thought to be associated with a polymorphism of the manganese superoxide gene (MnSOD) [37]. The gene locus of manganese superoxide dismutase has been mapped to the long arm of chromosome 6. Shimoda-Matsubayashi and her colleagues were unable to identify a pathogenic mutation [37]. However, this information provided important clues for the identification of a new causative gene for young-onset PD. In 1998, Kitada et al. [38] identified a novel causative gene, parkin, using the Keio BAC (bacterial artificial chromosome) library. The parkin cDNA consists of a ubiquitin-like domain and two Really Interesting New Gene (RING) finger motifs. As neuropathologic examination of patients with parkin mutations showed a lack of Lewy bodies, the group hypothesized that the function of this gene might be involved in proteolytic systems, such as the ubiquitin‒proteasome pathway. Subsequently, parkin was shown to encode the E3 ubiquitin ligase, which is directly linked to the ubiquitin‒proteasome system [39]. Parkin protein is involved in mitophagy, a type of autophagy, in collaboration with PTEN-induced putative kinase 1 (PINK1) [40]. This information suggests that both molecules share a common pathway, mitophagy, that is responsible for the removal of abnormal mitochondria.
- Parkin-mutation-induced astrocytic alteration, especially reduced astrocytosis, suggests the possibility of an astrocyte-related nonautonomous cell death mechanism for dopaminergic neurons in the brains of patients with parkin mutations [41]. Recent evidence has shown that PINK1-Parkin functions as the core machinery for mitophagy in not only neurons but also astrocytes, and deficiency in this pathway causes young-onset PD [42].
- A prominent clinical feature of parkin-linked PD, especially in very early-onset patients, is dystonia, with a prevalence of up to 85% in those with disease onset of 20 years or younger. Patients are especially susceptible to dopa-induced dyskinesia and motor function fluctuation (Supplementary Video 2 in the online-only Data Supplement) [38]. More widespread genetic testing of the parkin gene has widened the clinical spectrum. For example, Klein et al. [43] reported the presence of parkin deletions in a family with adult-onset, tremor-dominant parkinsonism. More recently, a “benign tremulous parkinsonism” phenotype was proposed [44]. The patients had juvenile-onset tremors for many years before the appearance of parkinsonism and had a dramatic and sustained response to anticholinergic monotherapy [44]. Initially, parkin-associated PD was thought to be peculiar to Japan; however, it is now known to be found throughout the world [45].
- Regarding treatment, most patients with parkin mutations respond favorably to levodopa treatment even at a low dose, albeit with a propensity to develop motor complications. In addition, it appears that deep brain stimulation (DBS) is a good option for parkin mutation carriers [46,47]. Recently, improvements in symptoms with apomorphine infusion has been reported [48].
- XDP; Lubag disease; DYT-TAF1; DYT3
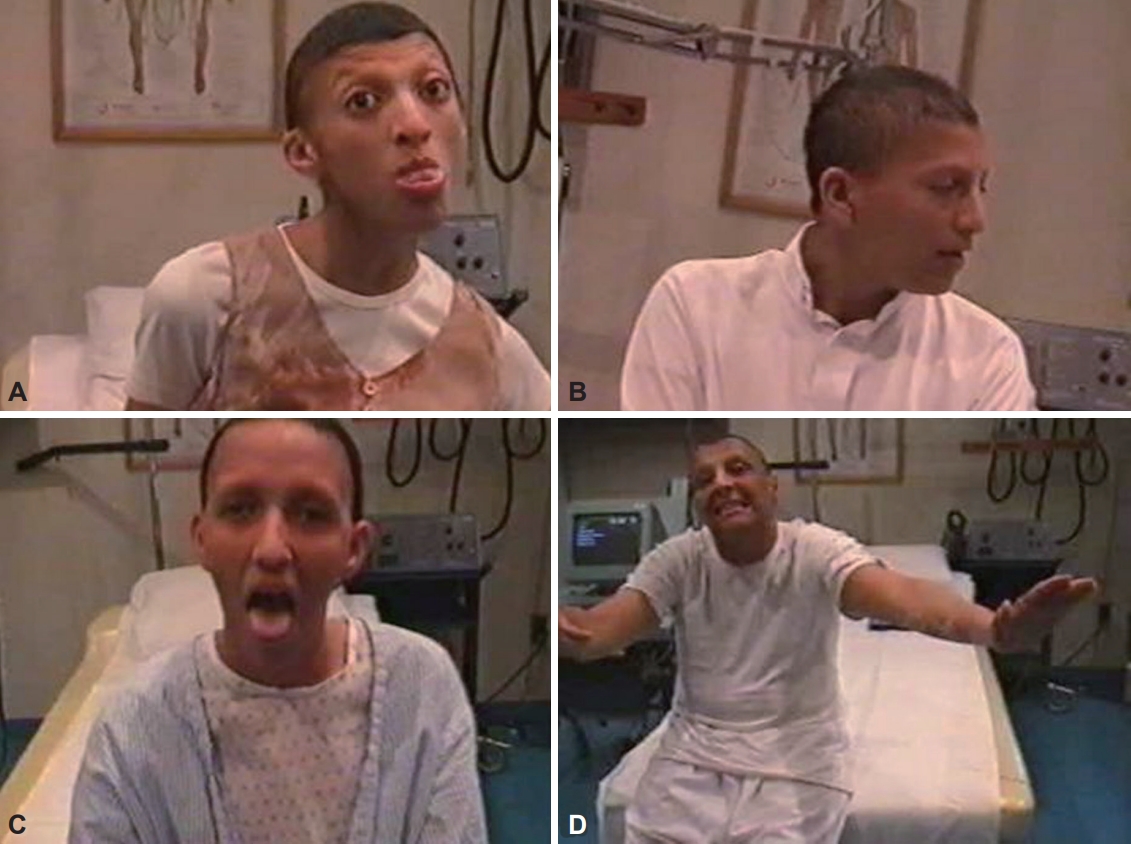
- XDP came to medical attention in 1969 when Dr. George Viterbo described the cases of five males labeled as having “dystonia musculorum deformans” from the Island of Panay [49,50]. This finding resulted in an epidemiological survey of patients on the island [51]. In 1975, 28 cases of “torsion dystonia of Panay” were presented at the Second Dystonia Meeting in New York City and, in 1976, were published in a landmark paper [49]. All patients were male. There was no male-to-male transmission, so sex-linked transmission was inferred, which was previously undescribed at that time [49]. Since then, an XDP Study Group and Registry has been formed in the Philippines, and an alliance with the local government and Health Department has been forged, with a system put in place to identify, refer and study suspected cases. After 25 years of follow-up, the natural history of the disease has become much clearer [50,51].
- The disease usually progresses in 3 phases. The disease starts with the dystonic phase, where patients typically present with focal dystonia that can involve any part of the body [50,51]. Focal dystonia generalizes in 84% of patients within 5 years [50]. Patients will usually have severe, nearly constant axial dystonia, retrocollis and prominent oromandibular dystonia, such as jaw opening and tongue protrusion (Supplementary Video 3 in the online-only Data Supplement). These features prevent the patients from assuming any comfortable position, but they can still move about in a bizarre fashion. This dystonic phase lasts between 2 and 7 years; however, as early as the 2nd year, parkinsonian features may start to appear. At the 7th to 10th year of illness, dystonia reduces, and parkinsonism becomes more prominent. This is referred to as the combined dystonia-parkinsonian phase. By the 15th year of illness, called the parkinsonian phase, the predominant picture is Parkinson-like. Not all patients reach this stage, as most patients die within 10 years. Less than 6% present with parkinsonism initially, who usually also have a later age at onset and a more benign course (Supplementary Video 4 in the online-only Data Supplement) [52,53]. In addition, non-motor features can significantly contribute to a patient’s disability. Cognition (including executive, attentional or visuospatial domains), mood, and sensory (pain) or autonomic (increased sweating) functions may be affected [52,54]. Most female XDP carriers are asymptomatic. Symptomatic females may have focal dystonia, chorea, focal tremors and parkinsonism, which are generally less severe. Detailed genealogical studies have traced almost all cases to mothers with roots on Panay Island.
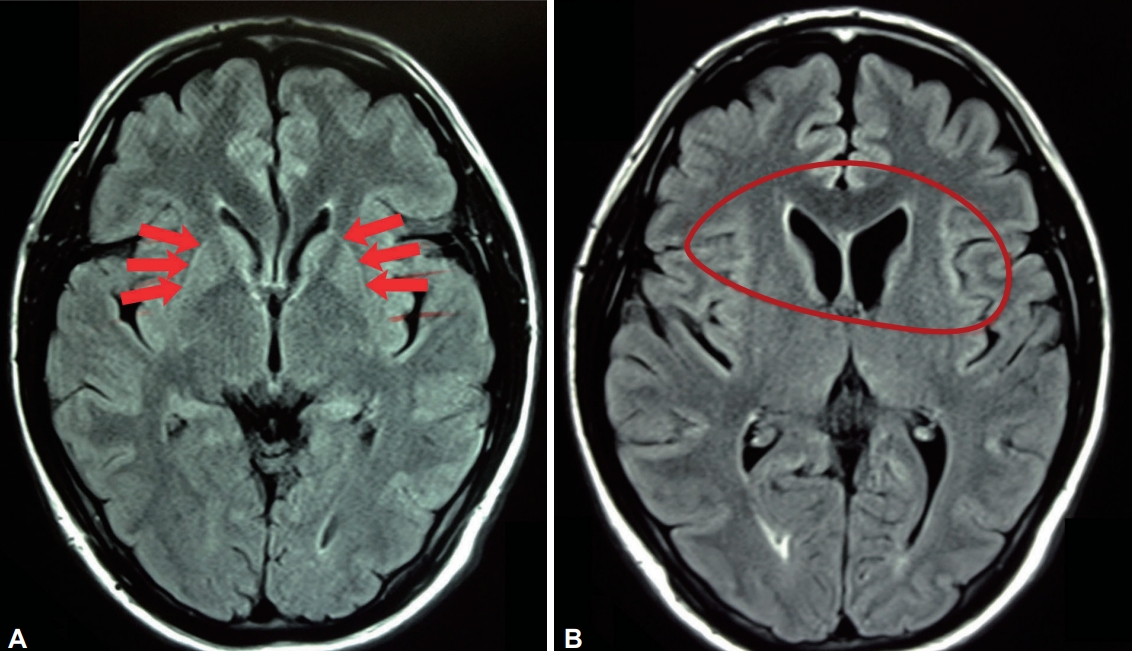
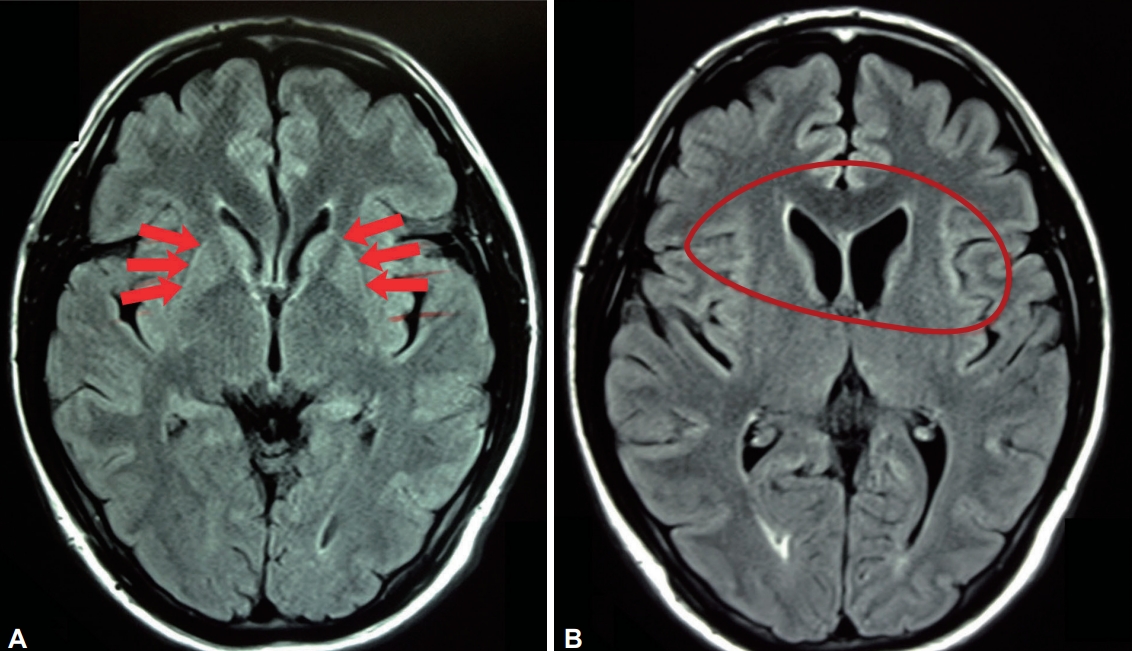
- Magnetic resonance imaging (MRI) findings of markedly hyperintense putaminal rims are a constant feature in all phases of the disease. Caudate head and putaminal atrophy are most prominent in the combined and predominantly parkinsonian stages [55]. White matter and pallidal abnormalities are also described (Figure 1) [56].
- X-linked inheritance was confirmed by subsequent linkage disequilibrium analysis narrowing the gene location to a < 350 kb region in Xq13.1 [57,58]. Nolte and his colleagues [59] described disease-specific changes (DSCs) and a potential multiple transcript system (MTS) in the distal portion of the Taf1 gene, and Makino et al. [60] reported SVA (short interspersed nuclear element, variable number of tandem repeats, and Alu composite) retrotransposon insertion in intron 32 of the Taf1 gene.
- The SVA element in XDP includes a hexameric DNA repeat expansion, (CCCTCT)n, the length of which was found to be inversely correlated with the age of disease onset [61,62]. Moreover, the longer the hexameric repeats, the more prominent the dystonic component, whereas shorter repeats were more frequently associated with parkinsonian features [61,62].
- In general, the response of XDP symptoms to oral medical therapy has been poor and inconsistent [63]. However, patients have shown improvements with regular botulinum toxin injections and bilateral pallidal DBS, with dystonia responding to DBS better than parkinsonism (Supplementary Video 5 in the online-only Data Supplement) [64-69].
- DRPLA
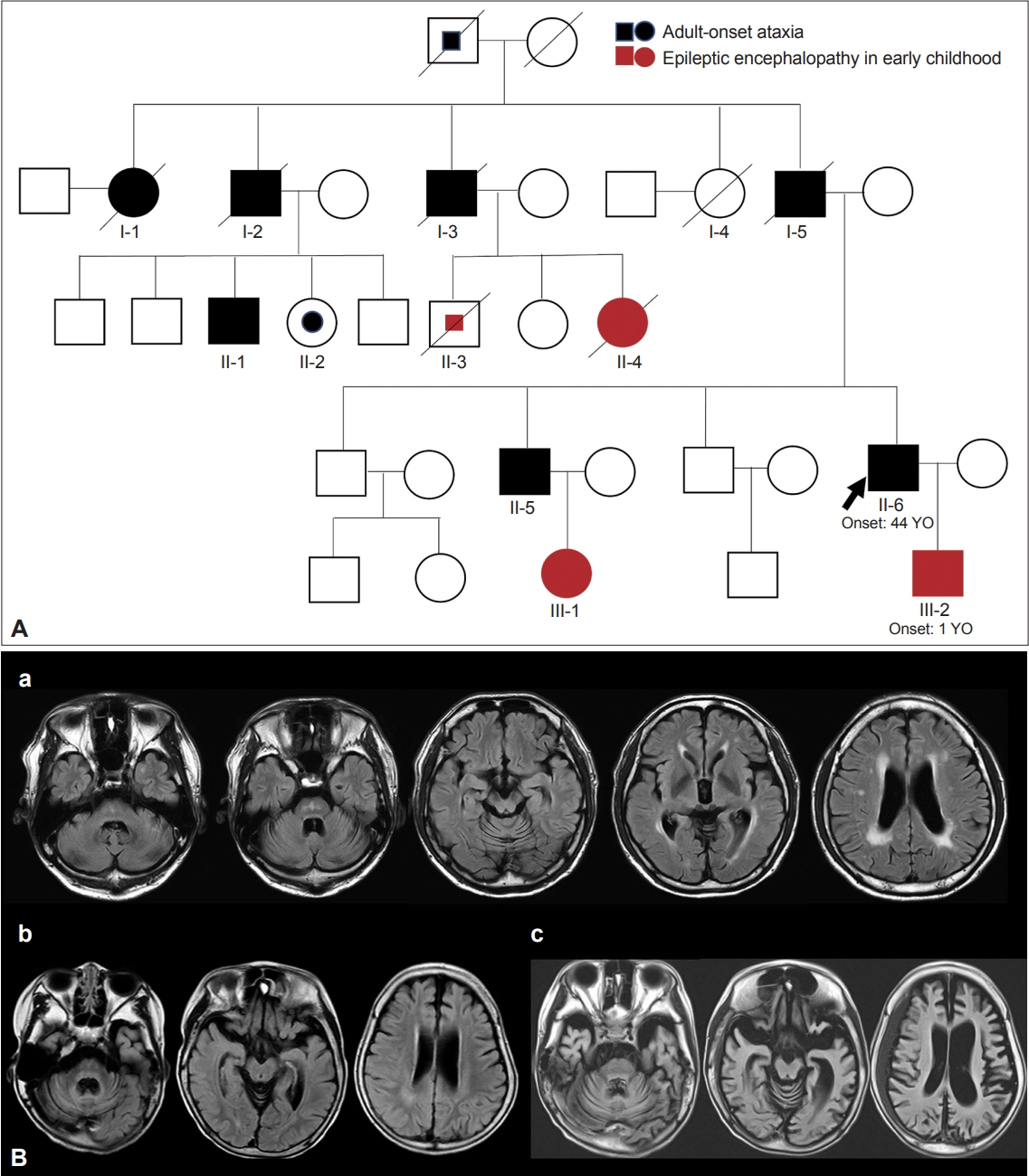
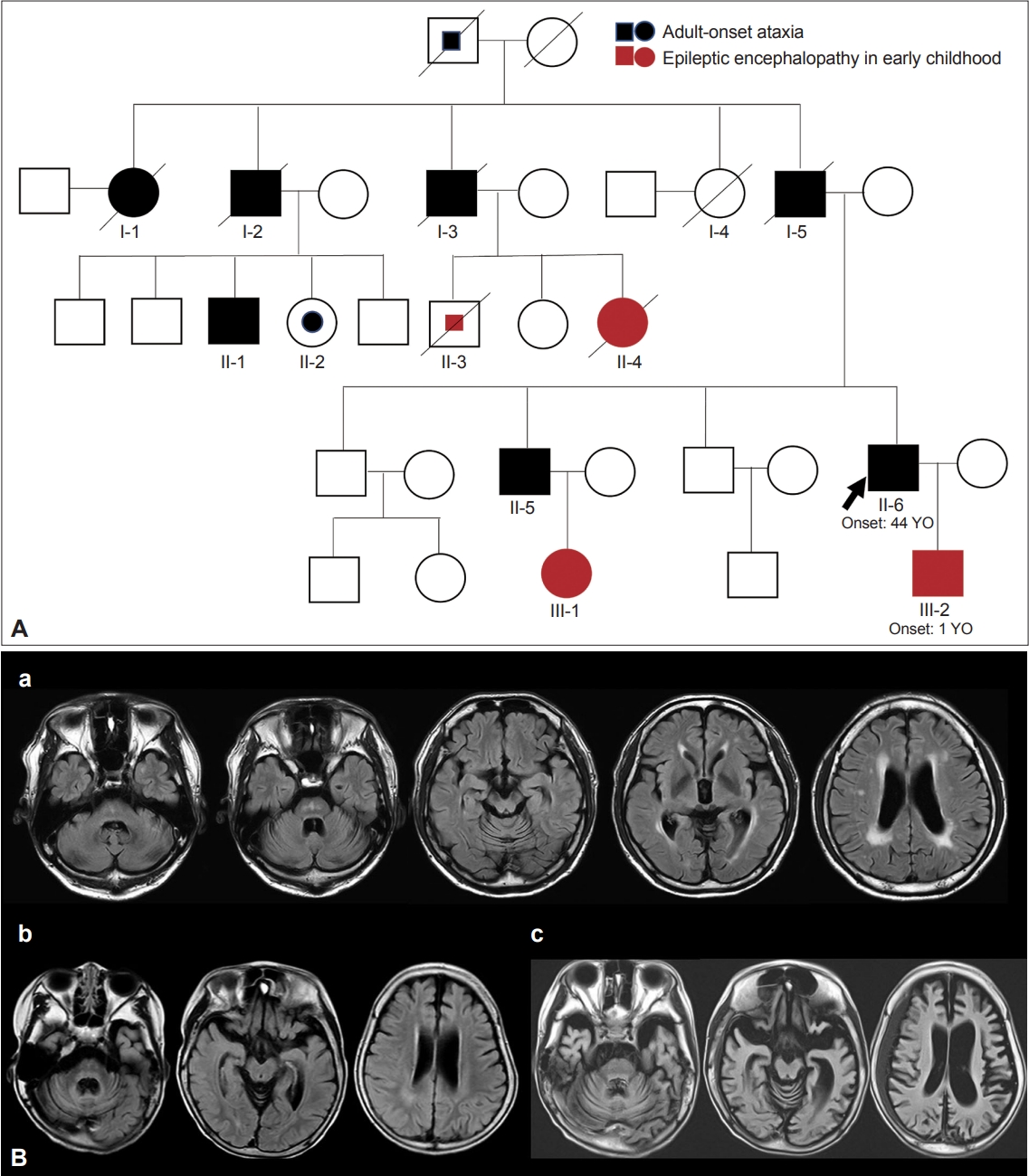
- The name DRPLA was first proposed by Smith et al. [70] in 1958. He reported the cases of patients with an unusual form of cerebellar ataxia associated with choreoathetosis whose pathological study showed the atrophy of the dentatorubral and pallidoluysian systems. The name was based on only the pathologically affected sites of lesions. Thereafter, several case reports of DRPLA were published in Japan based on only pathological findings. In 1972, 14 years later, Naito et al. [71] reported a dominantly inherited form of myoclonic epilepsy in Japanese individuals. It was at this time when the present disease entity of DRPLA, an autosomal dominantly inherited disease with epilepsy and ataxia, was established. Then, in 1978, the clinical picture from Japan was reported in an English journal [72]. In 1982, based on these accumulated clinical descriptions, the present disease concept of DRPLA was established by Naito and Oyanagi [73]. Their report fully described the clinical and pathological features of DRPLA, all of which are now accepted as typical features. These features comprise progressive myoclonic epilepsy, choreoathetosis, and dementia with an autosomal dominant form of inheritance. In some cases, mainly cerebellar ataxia or choreoathetosis (usually adult-onset) or epilepsy and dementia (usually childhood- or juvenile-onset) are shown in the same family, demonstrating intrafamilial heterogeneity (Figure 2 and Supplementary Video 6 in the online-only Data Supplement). In 1984, Iizuka et al. [74] also proposed the classification of DRPLA into three types based on their own experience and previously reported cases. Their concept is broader than the current DRPLA concept. It is assumed that some of these types included other diseases. In 1988, Takahashi et al. [75] demonstrated the common characteristics of combined degeneration of the dentatorubral and pallidoluysian systems and the pathological diversity of the disease. Viewing the process to establish the DRPLA concept, we can trace the amazing footsteps of Japanese clinicians and neuropathologists who, at a time when there were no search engines such as PubMed, carefully reviewed the literature to uncover unknown disease groups.
- DRPLA has been reported more frequently in individuals of Japanese ancestry and less frequently in individuals from other Asian countries, such as Singapore, Korea and China [76]. DRPLA is also reported less frequently in individuals of European ancestry [76]. In 1989, an African-American family living around the Haw River in North Carolina, USA, with a five-generation history of ataxia, seizures, chorea, progressive dementia, and death within 15 to 25 years, was reported as having “Haw River syndrome” [77]. In 1994, Haw River syndrome was confirmed to be DRPLA through genetic analysis [78].
- In 1990, Kondo et al. [79] showed that DRPLA is a disease distinct from Huntington disease by linkage analysis using a single marker. In the early 1990s, various neuromuscular diseases were determined to be caused by an increase in the number of repetitive sequences in the gene [80]. These discoveries revealed that repeat lengths are unstable from generation to generation and that there is a close relationship between clinical severity and repeat length. In June 1993, Li et al. [81] isolated several gene fragments that met the criteria for increased repetition, but they failed to find these repetitive sequences in genetic neuropsychiatric disorders. In 1994, two Japanese groups simultaneously found that the CAG repeat sequence in CTG-B37, reported by Li et al. [81], was increased in patients with DRPLA [82,83], leading to the identification of the gene for the disease. The results of the genetic analysis showed that the clinical and pathological variability between generations could be explained by the degree of repeated sequences. The exact function of the isolated gene and therapeutic methods have not yet been established. However, model mice carrying the full-length genomic sequence of human Atrophin-1 (the causative gene of DRPLA), including the promoter and the expanded CAG repeat sequence, have been created in Japan [84]. We hope that a treatment method for this disease will also be developed.
- WSS
- The name WSS is derived from the two doctors who, in 1983 [85], described the cases of six patients from two consanguineous Saudi Arabian families who had an adolescent-onset autosomal recessive disorder, with characteristic facial appearance, hypogonadism, mental abnormalities, alopecia or sparse hair, sensorineural deafness, diabetes mellitus and electrocardiogram (ECG) abnormalities (Figure 3). All female patients had failed sexual maturation. Males had testicular failure [85].
- Similar cases were subsequently described in Kuwait by AlAwadi et al. [86] in 1985 and in Turkey by Gül et al. [87] in 2000. Due to the lack of full phenotypic manifestations, the cases by Al-Awadi et al. [86] were not considered WSS by the authors at the time. In 2007, Al-Semari and Bohlega published an extensive phenotypic description and laboratory and MRI findings of WSS in 26 affected patients from 12 consanguineous families in Saudi Arabia, including the first six patients described by Woodhouse and Sakati [88]. In addition to alopecia, hypogonadism, deafness and diabetes mellitus, 17 patients developed either progressive generalized or focal dystonia. All patients had low insulin-like growth factor 1 (IGF-1) levels. Brain MRI showed low signal intensities in the basal ganglia with white matter hyperintensities.
- WSS was concluded to be most likely a multisystemic disorder and may in fact have been described earlier by Crandall et al. [89] in 1973 in three siblings presenting with alopecia, hypogonadism and sensorineural deafness, prior to the description by Woodhouse and Sakati [85], and may also include the syndrome described by Al-Awadi et al. [86] WSS has since been described in other ethnic groups, including Italian [90], Indian [91], Turkish [87], Pakistani [92], Tunisian [93] and Portuguese ethnicities [94], with variable and less severe phenotypes as well as variable neuroimaging features. More recently, a detailed report of an additional 58 patients from Qatar [95] was published suggesting that WSS is perhaps more common among Arab individuals and may be a reflection of the endogamous marital practices of the Arabic community.
- A comprehensive review in 2008 categorized WSS features into endocrine manifestations (alopecia, hypogonadism, diabetes mellitus and thyroid dysfunction), progressive neurological syndrome (dystonia, mental retardation, sensorineural deafness) and unspecified abnormalities such as ECG abnormalities, keratoconus, camptodactyly and acanthosis nigricans [96].
- In a recent review, two distinct phenotypic spectra were reported [97]. Type 1 had onset in early adolescence, a more disabling and rapidly progressive disease course, and more frequent occurrence of intellectual disability (Supplementary Video 7 in the online-only Data Supplement). Type 2 presented in late adolescence with a milder disease course, less severe disability, and relatively preserved intellectual abilities (Supplementary Video 8 in the online-only Data Supplement). Neurological involvement was present in 81.5% of these patients, with dystonia being the most common associated symptom, followed by mental impairment and sensorineural deafness in 30% [97].
- The causative mutation was identified in 2008 as a single base pair deletion (c.436delC) in the gene C2orf37, now known as DCAF17, which encodes a nucleolar protein [98]. Mutations causing a single base pair deletion in exon 3 of the DCAF17 gene were discovered to be the cause of WSS in Pakistani families [99]. WSS is characterized by variable interfamilial and intrafamilial phenotypic heterogeneity, including a milder phenotype with normal intellectual abilities and an absence of extrapyramidal syndrome. Similarly, other phenotypes of WSS caused by the C2orf37 mutation were described in seven patients from Qatar, in which patients presented without diabetes mellitus, sensorineural hearing loss or dystonia [99].
- Common brain imaging findings of WSS include a small pituitary gland, iron deposition in the globus pallidus and nonenhancing white matter hyperintensities in the periventricular and frontoparietal regions (Figure 4). Less common findings include restricted diffusion in the splenium of the corpus callosum and prominent perivascular spaces [100]. To date, more than 122 cases have been described, mostly affecting Arab families [95,101,102].
- BAFME
- BAFME is also known as familial adult myoclonic epilepsy (FAME); familial cortical myoclonic tremor with epilepsy (FCMTE); and autosomal dominant cortical tremors, myoclonus, and epilepsy (ADCME).
- The cases of patients considered to be affected by BAFME have been reported in Japan before the establishment of the clinical entity of BAFME in the 1990s based on familial, nonprogressive epileptic clinical features and autosomal dominant inheritance [103-105]. Case reports can be traced back to the 1920s (summarized in Inazuki 1990) [106,107]. In 1979, Naito and Kaji [108] tried to classify the conditions of 169 individuals from 39 families with dominantly inherited myoclonus epilepsy and identified that approximately 15%–20% of affected patients had adult-onset, non-progressive clinical features. Inazuki et al. [106] summarized the detailed clinical features in 1990. In the same year, Ikeda et al. [109] revealed giant somatosensory evoked potential (SEP) recorded on the contralateral primary sensory cortex and C reflex after the stimulation of the median nerve. They also detected myoclonus-related cortical activity by the jerk-locked backaveraging method. They named the involuntary movement cortical tremors and concluded that these tremors represented a variant of cortical reflex myoclonus [109]. Yasuda independently described their findings from a detailed clinical and electrophysiological study of large families in 1991 [110].
- Currently, the accepted core clinical features of BAFME are as follows [111]: 1) an autosomal dominant mode of inheritance with high penetrance, 2) minimally progressive cortical tremors (tremulous myoclonus) (Supplementary Video 9 in the online-only Data Supplement) and infrequent seizures, and 3) electrophysiological study findings of cortical hyperexcitability underlying the cortical tremors. The onset of cortical tremors often precedes the onset of seizures. Whereas the disease is sometimes recognized as virtually non-progressive, there are electrophysiological studies showing progression by age [112,113].
- Genetic linkage studies were conducted, and most affected Japanese families and an affected Chinese family was shown to be linked to 8q24 (BAFME1) [114-118]. Outside East Asia, similar diseases have been identified, and linkage analysis revealed additional independent loci on the centromeric region of chromosome 2 (BAFME2) [119-124], chromosome 5 (BAFME3) [125], and chromosome 3 (BAFME4) [126]. In 2018, Ishiura et al. [127] discovered pentanucleotide repeat expansions in intron 4 of SAMD12 as the cause of BAFME1. The lengths of the repeat expansions and their instability over generations explained the anticipation phenomenon observed in BAFME families [127,128]. The pathogenic repeat expansions include expansions of TTTTA and TTTCA repeats, while a short TTTTA repeat is registered in the human reference sequence. In situ hybridization analysis revealed RNA foci containing UUUCA repeats in autopsied brains [127]. These observations raised the possibility that an RNA-mediated mechanism, particularly by UUUCA repeats, is strongly involved in the pathogenesis of BAFME [127]. The study also revealed two families without repeat expansions in SAMD12; instead, the researchers identified repeat expansions including TTTCA and TTTTA repeats in the introns of TNRC6A and RAPGEF2 in these families and designated the diseases as BAFME6 and BAFME7, respectively [127]. BAFME1 is also found in Sri Lanka, India, and Thailand [129-131]. Founder haplotypes were identified in the families [129,131,132], suggesting that repeat expansions rarely occurred in human history.
- After the discovery of pentanucleotide repeats in three genes in BAFME1, BAFME6, and BAFME7, BAFMEs linked to chromosomes 2, 3, and 5 were discovered to be caused by expansions of TTTCA and TTTTA repeats in STARD7 (BAFME2) [133], YEATS2 (BAFME4) [134], and MARCHF6 (BAFME3) [135], respectively, as predicted previously [127]. These findings emphasize a strong repeat motif-phenotype correlation in BAFME [136]. This finding that expansions of identical repeat motifs lead to similar diseases strongly supports the theory that the expression of RNA molecules containing these repeats is involved in the pathogenesis of BAFME.
- Patients with BAFME are usually treated with anticonvulsants such as sodium valproate, clonazepam, levetiracetam, piracetam, zonisamide, and primidone. Epilepsy in BAFME is generally easily controlled. The effect of low-dose perampanel on cortical tremors was also reported recently [137].
- Kufor-Rakeb syndrome; PARK-ATP13A2; PARK9
- Kufor-Rakeb syndrome was first described by Najim Al-Din et al. [138] in 1994 as a juvenile-onset, autosomal recessive, progressive pallido-pyramidal syndrome in a Jordanian family. In November 1992, a patient from the Kufor-Rakeb village in northern Jordan visited his clinic with unique clinical features. Najim Al-Din and two other doctors visited the community and examined 87 family members. Of these, five siblings born to consanguineous parents had neurological symptoms. All of them had normal birth histories and development milestones until the end of the 1st decade, when they started to develop slowness of movement, predominantly affecting their walking at approximately 13 years (range 12–16 years). The older two affected patients had poor intellectual ability. All had typical mask-like facies and developed progressive supranuclear gaze palsy and moderate to severe corticospinal tract involvement, in addition to marked parkinsonism (akinetic-rigid type, without tremors). The disease was described to be rapidly progressive, and the patients were bedridden within 2 years [138]. Patients were started on levodopa, which resulted in dramatic improvement in parkinsonism and improvement in walking in the younger siblings [138]. The cognitive difficulties, spasticity and supranuclear gaze palsy progressed over time, with the eventual development of levodopa-induced dyskinesia associated with narrowing of the therapeutic window, cognitive decline, and the appearance of facial-faucial mini myoclonus and visual hallucinations [139]. Brain MRI showed pallidal, pyramidal and generalized cerebral atrophy [138].
- In 2001, in a genetic linkage analysis study of the Jordanian family performed by Hampshire et al. [140], the syndrome was mapped to the 9 cM region of chromosome 1p36, which was assigned as PARK9. The gene responsible was later identified by Ramirez et al. [141] as the ATP13A2 gene encoding lysosomal type 5-P type ATPase through genetic analysis of a large Chilean kindred presenting with autosomal recessive juvenile-onset parkinsonism and dementia similar to the originally described KuforRakeb syndrome. This mutation in the ATP13A2 gene was also verified in the original Jordanian family [141]. In the Chilean kindred, symptoms were observed in compound heterozygote carriers of ATP13A2 gene mutations, and there were T2-weighted hypointensities in the caudate and lenticular nuclei on brain MRI, suggestive of brain iron accumulation [142]. The Chilean family had 2 different missense mutations (one in the father and one in the mother), whereas in the Jordanian family, there was a 22-bp duplication in all affected family members leading to a frameshift and stop codon after 236 extraneous amino acids leading to a loss of function [141]. Since then, other phenotypes have been described in other populations, including juvenile-onset parkinsonism [143], ataxia-myoclonus [144], action myoclonus and seizures [145], ataxia and polyneuropathy [146], neuronal ceroid lipofuscinosis type 12 [147], complicated hereditary spastic paraplegia type 78 (HSP78) [148], juvenile-onset amyotrophic lateral sclerosis [149] and prominent psychiatric and behavioral manifestations [150]. The ATP13A2 gene mutation is now included as a rare cause of dystonia-parkinsonism151 and neurodegeneration with brain iron accumulation (Supplementary Video 10 in the online-only Data Supplement) [151,152].
- CAMTA2 tremulous dystonia; Bohlega syndrome
- This syndrome was first described by Bohlega et al. [153] in 1995 in two unrelated Saudi Arabian families, with a clinical presentation of juvenile onset, myoclonic tremor syndrome, and spasticity associated with cerebral white matter changes. The disease is autosomal recessive and has an onset age in the first decade of life. In the first family, the syndrome began at an average age of 7 years, with a slow frequency tremor (4–6 Hz) affecting the arms at rest. The arm tremor was worsened by posture and action, and there was a side-to-side (no-no) head tremor associated with focal or segmental craniocervical dystonia. In the second family, the tremor was jerky and involved both hands, with mild orolingual and facial twitches, consistent with myoclonus. In both families, the disease was slowly progressive, with the presence of spasticity later in the disease course. Patients were followed up for 24 years with no change in the movement disorder, there were no changes in cognition or intellectual ability, and patients were able to lead a relatively normal life, except for some mild gait difficulties later in adulthood that were due to spasticity (Supplementary Video 11 in the online-only Data Supplement). Accelerometric recording from an outstretched hand showed 4-Hz tremors, and brain MRI showed bilateral confluent white matter changes not characteristic of any of the classic leukodystrophies and with no change over time [153,154]. The results of screening for lysosomal and metabolic enzyme deficiencies, including arylsulfatase levels for metachromatic leukodystrophy, were negative in both families.
- Recently, in 2017, the candidate gene underlying this unique dystonia syndrome proposed by the authors as “Bohlega syndrome” was identified as the CAMTA2 (Calmodulin Binding Transcription Activator 2) gene, encoding the CAMTA2 protein, a member of the calmodulin—binding transcription activator protein family. A common domain structure is shared in this family of proteins, including a transcription activation domain, a DNA-binding domain, and a calmodulin-binding domain [154]. CAMTA2 has six protein-coding isoforms (CAMTA2 ENSG00000108509) ranging from 178 to 1241 amino acids; however, the expression pattern and function of each isoform remain unknown [154]. CAMTA2 is expressed in the heart and brain. Diseases associated with CAMTA2 include retinitis pigmentosa and cardiac valvular dysplasia [154]. At this time, there have been no new reports of this syndrome in any other populations outside these Saudi Arabian families. Further replication studies with large dystonia cohorts in other ethnicities are needed to confirm the role of CAMTA2 in patients with dystonia.
- PKD; PxMD-PRRT2; PKD-PRRT2
- ‘Familial paroxysmal choreoathetosis’ was first proposed by Mount and Reback [155] to describe the condition of a 23-year-old white man, who showed infantile-onset intermittent choreodystonic attacks that lasted several hours and could be induced by alcohol, coffee, and fatigue, and several of his affected family members in an autosomal dominant pattern of inheritance. He probably referred to what we now know as paroxysmal non-kinesigenic dyskinesia (PNKD). Andrew Kertesz, in 1967, introduced the term “kinesigenic”, which means “induced by movement” [156]. He highlighted the differences between the cases where patients had short attacks precipitated by movements, thus differentiating them from the cases described by Mount and Reback [156]. In 1973, Toyokura discovered that Shuzo Kure, a Japanese psychiatrist, had originally described the disorder that is now termed PKD in 1892 [157].
- The original description, supported by hand-traced high-speed photographs (Figure 5), to be the first case of PKD was reported in the Japanese literature in 1892 by Shuzo Kure, who provided details of the attacks in a 23-year-old man that were triggered by sudden movement that brought an odd sensation as a kind of sensory aura that was rapidly followed by peculiar, purposeless, irregular involuntary movements of the legs of a very short duration, sometimes spreading to the right side of the body [157,158]. Although Kure labeled the condition as atypical Thomsen’s disease (myotonia congenita), noting the lack of long-lasting muscle contractions during the attack and percussion myotonia (hence, “atypical”), his clinical descriptions on the clinical manifestations as we currently recognize as PKD remain valid, except for a lack of being hereditary. In His image showed a chair placed behind the patient denoting attacks brought about by quickly rising from the chair (Figure 5) [157,158].
- Studies have recognized several clinical manifestations of paroxysmal dyskinesias that are inclusive of dystonia, chorea, athetosis, ballism, or any form of dyskinesias that may occur in isolation or as a combination and suddenly develop after precipitants of attacks of a variable duration, resulting in the most widely adopted classification into three subtypes based on triggering factors, namely, PKD, PNKD and paroxysmal exercise-induced dyskinesia (PED) [159,160]. Further genetic discoveries in these subtypes, where mutations of the PRRT2 [161,162], PNKD or MR-1 [163,164], and SLC2A1 [165] genes account for the major primary forms of PKD, PNKD, and PED cases, respectively, have expanded our understanding that there is a marked pleiotropy of mutations in such genes with a still expanding clinical spectrum (i.e., hemiplegic migraine, ataxia, and myotonia in complicated PKD), and not all patients clinically presenting with PKD, PNKD, or PED have mutations in these genes [166]. PRRT2 mutations can be associated with a spectrum of disorders from benign familial infantile epilepsy and infantile convulsion to PKD. This and the prevalence of PKD, suggesting that it is more common in Asians, have been reviewed elsewhere by the TF [167].
RESULTS
- These nine conditions first described in Asia continue to hold great value for all generations of neurologists. We can all learn how these astute clinicians made their observations and how these observations triggered advances that have led to new knowledge and therapies of the underlying disorders. The original authors showed admirable clinical curiosity, taking their observations further and not ignoring the chance observations or unexplained phenomena. Obviously, these discoveries were not based on a single outpatient visit but rather on a series of longitudinal observations that were supplemented by information from the surroundings, such as family members, and backed up by subsequent genetic investigation, community or epidemiological studies and an alliance with government bodies in certain cases. However, in our view, this series of original contributions provides timeless pieces of work that set a good example to aspiring clinicians in this and next generations to encourage their sense of originality and continual curiosity to pursue what they cannot explain.
CONCLUSION
Supplementary Material
Video 1.
Video 2.
Video 3.
Video 4.
Video 5.
Video 6.
Video 7.
Video 8.
Video 9.
Video 10.
Video 11.
-
Ethics Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. Written informed consent from patients was obtained for all the supplementary videos and figures with patient identification materials except where stated in the legend.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None
-
Author contributions
Conceptualization: Priya Jagota, Yoshikazu Ugawa, Zakiyah Aldaajani, Norlinah Mohamed Ibrahim, Cid Diesta, Shen-Yang Lim, Jee-Young Lee, Beomseok Jeon, Pramod Kumar Pal, Huifang Shang, Shinsuke Fujioka, Prashanth Lingappa Kukkle, Onanong Phokaewvarangkul, Chin-Hsien Lin, Cholpon Shambetova, Roongroj Bhidayasiri. Methodology: Priya Jagota, Yoshikazu Ugawa, Zakiyah Aldaajani, Norlinah Mohamed Ibrahim, Cid Diesta, Shen-Yang Lim, Jee-Young Lee, Beomseok Jeon, Pramod Kumar Pal, Huifang Shang, Shinsuke Fujioka, Prashanth Lingappa Kukkle, Onanong Phokaewvarangkul, Chin-Hsien Lin, Cholpon Shambetova, Roongroj Bhidayasiri. Writing—original draft: Priya Jagota, Yoshikazu Ugawa, Zakiyah Aldaajani, Norlinah Mohamed Ibrahim, Hiroyuki Ishiura, Yoshiko Nomura, Shoji Tsuji, Cid Diesta, Nobutaka Hattori, Osamu Onodera, Roongroj Bhidayasiri. Writing—review & editing: all authors.
Notes
- We would like to thank Prof. Raymond L. Rosales (MD, PhD, The Research Center for Health Sciences, Faculty of Medicine & Surgery, University of Santo Tomas, Manila, Philippines), Assoc. Prof. Roopa Rajan (MD, Department of Neurology, All India Institute of Medical Sciences, New Delhi, India), and Prof. Arlene R. Ng (MD, Institute for Neurosciences, St. Luke’s Medical Center Global City, Taguig City, Philippines), for providing their opinions, figures and supplementary videos.
Acknowledgments


| Disorder | Genes | Inheritance | Original report | Author(s) | Country | Main clinical features |
|---|---|---|---|---|---|---|
| Segawa disease; DYT/PARK-GCH1; DYT5a | GTP cyclohydrolase1 (GCH1) | AD | Childhood basal ganglia disease with remarkable response to L-DOPA, “hereditary basal ganglia disease with marked diurnal fluctuation”. Shinryo (Therapy-Tokyo) 1971;24:667-672 | Segawa et al. [9] | Japan | Dystonia, usually starting in the lower limbs, with marked diurnal variation. Responsive to low-dose levodopa |
| PARK-Parkin; PARK2 | Parkin | AR | Four cases of juvenile paralysis agitans in a family. Psychiatr Neurol Jap 1958;60:178-186 | Nasu et al. [33] | Japan | Early-onset dystonia and parkinsonism with high propensity for motor fluctuation |
| X-linked dystonia parkinsonism (XDP); Lubag disease; DYT-TAF1; DYT3 | Taf1 | X-linked | Torsion dystonia in Panay, Philippines. Adv Neurol 1976;14:137-151 | Lee et al. [49] | Philippines | Early-onset generalized dystonia with prominent and severe oromandibular and axial dystonia |
| Dentatorubral-pallidoluysian atrophy (DRPLA) | Atrophin-1 | AD | Two families of progressive myoclonus epilepsy with Mendelian dominant heredity. Seishin Shinkeigaku Zasshi 1972;74:871-897 | Naito et al. [71] | Japan | Progressive myoclonic epilepsy, choreoathetosis, ataxia and dementia |
| Woodhouse-Sakati syndrome (WSS) | DCAF17 | AR | A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet 1983;20:216-219 | Woodhouse and Sakati [85] | Saudi Arabia | Progressive generalized or focal dystonia, intellectual impairment, alopecia, hypogonadism, deafness and diabetes mellitus |
| Benign adult familial myoclonic epilepsy (BAFME) | SAMD12 (BAFME1), STARD7 (BAFME2), MARCHF6 (BAFME3), YEATS2 (BAFME4), TNRC6A (BAFME6), RAPGEF2 (BAFME7) | AD | A hereditary neurological disease presenting a kind of tremor and epileptic attacks resembling Unverricht–Lundborg’s myoclonus epilepsy. Psychiat Neurol Jpn 1924;24:484-495 | Nakazawa [107] | Japan | Minimally progressive cortical tremor (tremulous myoclonus) with infrequent seizures and electrophysiological studies demonstrating cortical hyperexcitability underlying the cortical tremor |
| Kufor-Rakeb syndrome; PARK-ATP13A2; PARK9 | ATP13A2 | AR | Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 1994;89:347-352 | Najim Al-Din et al. [138] | Jordan | Juvenile-onset dystonia-parkinsonism, progressive supranuclear gaze palsy, corticospinal tract involvement, facial-faucial mini myoclonus and cognitive decline |
| CAMTA2 tremulous dystonia; Bohlega syndrome | CAMTA2 | AR | Familial tremulous and myoclonic dystonia with white matter changes in brain magnetic resonance imaging. Mov Disord 1995;10:513-517 | Bohlega et al. [153] | Saudi Arabia | juvenile-onset myoclonic tremor and spasticity, associated with cerebral white matter changes |
| Paroxysmal kinesigenic dyskinesia (PKD); PxMD-PRRT2; PKD-PRRT2 | PRRT2 | AD | Atypical Thomsen’s disease. Tokyo Igakukai Zasshi (J Tokyo Med Assoc) 1892;6:505-514 | Kure [158] | Japan | Episodes of dyskinesia of short duration triggered by sudden movement |
- 1. Parkinson J. An essay on the shaking palsy. London: Whittingham and Rowland for Sheerwood, Neely and Jones; 1817.
- 2. Quinn N. Multiple system atrophy--the nature of the beast. J Neurol Neurosurg Psychiatry 1989;Suppl:78–89.ArticlePubMedPMC
- 3. Magalhães M, Wenning GK, Daniel SE, Quinn NP. Autonomic dysfunction in pathologically confirmed multiple system atrophy and idiopathic Parkinson’s disease--a retrospective comparison. Acta Neurol Scand 1995;91:98–102.ArticlePubMed
- 4. Kollensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al. Red flags for multiple system atrophy. Mov Disord 2008;23:1093–1099.ArticlePubMed
- 5. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 6. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015;30:1591–1601.ArticlePubMed
- 7. Cortelli P, Calandra-Buonaura G, Benarroch EE, Giannini G, Iranzo A, Low PA, et al. Stridor in multiple system atrophy: consensus statement on diagnosis, prognosis, and treatment. Neurology 2019;93:630–639.ArticlePubMedPMC
- 8. Wenning GK, Stankovic I, Vignatelli L, Fanciulli A, Calandra-Buonaura G, Seppi K, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord 2022;37:1131–1148.ArticlePubMedPMCPDF
- 9. Segawa M, Ohmi K, Itoh S, Aoyama M, Hayakawa H. [Childhood basal ganglia disease with remarkable response to L-DOPA, “hereditary basal ganglia disease with marked diurnal fluctuation”.]. Shinryo (Therapy-Tokyo) 1971;24:667–672.Japanese.
- 10. Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol 1976;14:215–233.ArticlePubMed
- 11. Segawa M, Hoshino K, Hachimori K, Nishiyama N, Nomura Y. A single gene for dystonia involves both or either of the two striatal pathways. In: Nicholson LFB, Faull RLM, editors. The Basal Ganglia VII. Vol 52. Bostron: Springer; 2002:155–163.
- 12. Segawa M. [Hereditary progressive dystonia (HPD) with marked diurnal fluctuation]. Adv Neurol Sci (Tokyo) 1981;25:73–81.Japanese.
- 13. Segawa M, Nomura Y, Kase M. Diurnally fluctuating hereditary progressive dystonia. In: Vinken PJ, Bruyn GW, Klawans HL, editors. Handbook of Clinical Neurology. Amsterdam: ElsevierScience Publishers; 1986:529–539.
- 14. Segawa M, Nomura Y. Hereditary progressive dystonia with marked diurnal fluctuation. In: Segawa M, editor. Hereditary progressive dystonia with marked diurnal fluctuation. Lancs, New York: Parthenon Publishing Group; 1993:3–19.
- 15. Segawa M, Nomura Y. Hereditary progressive dystonia with marked diurnal fluctuation. Pathophysiological importance of the age of onset. Adv Neurol 1993;60:568–576.PubMed
- 16. Segawa M, Nomura Y. Hereditary progressive dystonia with marked diurnal fluctuation and dopa-responsive dystonia: pathognomonic clinical features. In: Segawa M, Nomura Y, editors. Age-related Dopamine-Dependent Disorders. Basel: Karger; 1995:10–24.
- 17. McGeer E, McGeer P. Some characteristics of brain tyrosine hydroxylase. In: Mandel AJ, editor. New Concepts in Neurotransmitter Regulation. Boston: Spinger; 1973:53–68.
- 18. Steinfels GF, Heym J, Strecker RE, Jacobs BL. Behavioral correlates of dopaminergic unit activity in freely moving cats. Brain Res 1983;258:217–228.ArticlePubMed
- 19. Segawa M. Development of the nigrostriatal dopamine neuron and the pathways in the basal ganglia. Brain Dev 2000;22 Suppl 1:S1–S4.ArticlePubMed
- 20. Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 2003;54 Suppl 6:S32–S45.ArticlePubMed
- 21. Segawa M, Nomura Y. Nishiyama N. Dopa-responsive dystonia. In: Stacy MA, editor. Handbook of Dystonia. 1st ed. New York: CRC Press; 2006:219–243.
- 22. Segawa M, Nomura Y, Hayashi M. Dopa-responsive dystonia is caused by particular impairment of nigrostriatal dopamine neurons different from those involved in Parkinson disease: evidence observed in studies on Segawa disease. Neuropediatrics 2013;44:61–66.PubMed
- 23. Kreiss DS, Anderson LA, Walters JR. Apomorphine and dopamine D(1) receptor agonists increase the firing rates of subthalamic nucleus neurons. Neuroscience 1996;72:863–876.ArticlePubMed
- 24. Rajput AH, Gibb WR, Zhong XH, Shannak KS, Kish S, Chang LG, et al. Dopa-responsive dystonia: pathological and biochemical observations in a case. Ann Neurol 1994;35:396–402.ArticlePubMed
- 25. Hornykiewicz O. Striatal dopamine in dopa-responsive dystonia. Comparison, with idiopathic Parkinson’s disease and other dopamine-dependent disorders. In: Segawa M, Nomura Y, editors. Age-related Dopamine-Dependent Disorders. Basel: Karger; 1995:101–108.
- 26. Fujita S, Shintaku H. Pathogenesis and Abnormal Pteridine Metabolism in Hereditary Progressive Dystonia (HPD) with Marked Diurnal Fluctuation. Journal of Kushiro City General Hospital 1990;2:64–67.
- 27. Furukawa Y, Nishi K, Kondo T, Mizuno Y, Narabayashi H. CSF biopterin levels and clinical features of patients with juvenile parkinsonism. Adv Neurol 1993;60:562–567.PubMed
- 28. Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994;8:236–242.ArticlePubMedPDF
- 29. Lee WW, Jeon B, Kim R. Expanding the spectrum of Dopa-responsive dystonia (DRD) and proposal for new definition: DRD, DRD-plus, and DRD look-alike. J Korean Med Sci 2018;33:e184.ArticlePubMedPMCPDF
- 30. Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson’s disease in GTP cyclohydrolase 1 mutation carriers. Brain 2014;137(Pt 9):2480–2492.ArticlePubMedPMC
- 31. Kase M. Pitfalls in neurological disorders. Jpn Med J 1978;2850:3–11.
- 32. LeWitt PA, Newman RP, Miller LP, Lovenberg W, Eldridge R. Treatment of dystonia with tetrahydrobiopterin. N Engl J Med 1983;308:157–158.Article
- 33. Nasu H, Aoyama T, Morisada A. [Four cases of juvenile paralysis agitans in a family]. Psychiatr Neurol Jap 1958;60:178–186.Japanese.
- 34. Ota Y, Miyoshi S, Ueda O, Mukai T, Maeda A. Familial paralysis agitans juvenilis; a clinical, anatomical and genetic study. Folia Psychiatr Neurol Jap 1958;12:112–121.
- 35. Yamamura Y, Iida M, Ando K, Sobue I. A juvenile familial disorder with rigidospasticity, bradykinesia and minor dystonia alleviated after sleep. Clin Neurol 1968;8:233–243.
- 36. Yamamura Y, Sobue I, Ando K, Iida M, Yanagi T. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 1973;23:239–244.ArticlePubMed
- 37. Shimoda-Matsubayashi S, Matsumine H, Kobayashi T, Nakagawa-Hattori Y, Shimizu Y, Mizuno Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson’s disease. Biochem Biophys Res Commun 1996;226:561–565.ArticlePubMed
- 38. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608.ArticlePubMedPDF
- 39. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000;25:302–305.ArticlePubMedPDF
- 40. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803.ArticlePubMedPMCPDF
- 41. Kano M, Takanashi M, Oyama G, Yoritaka A, Hatano T, Shiba-Fukushima K, et al. Reduced astrocytic reactivity in human brains and midbrain organoids with PRKN mutations. NPJ Parkinsons Dis 2020;6:33.ArticlePubMedPMCPDF
- 42. Morales I, Sanchez A, Puertas-Avendaño R, Rodriguez-Sabate C, PerezBarreto A, Rodriguez M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020;68:2277–2299.ArticlePubMedPDF
- 43. Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, et al. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 2000;48:65–71.ArticlePubMed
- 44. Mulroy E, Balint B, Menozzi E, Latorre A, Bhatia KP. Benign tremulous parkinsonism of the young-consider Parkin. Parkinsonism Relat Disord 2019;65:270–271.ArticlePubMed
- 45. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 2018;33:730–741.ArticlePubMedPDF
- 46. Kim HJ, Yun JY, Kim YE, Lee JY, Kim HJ, Kim JY, et al. Parkin mutation and deep brain stimulation outcome. J Clin Neurosci 2014;21:107–110.ArticlePubMed
- 47. Rizzone MG, Martone T, Balestrino R, Lopiano L. Genetic background and outcome of deep brain stimulation in Parkinson’s disease. Parkinsonism Relat Disord 2019;64:8–19.ArticlePubMed
- 48. Lim SY, Ahmad-Annuar A, Lohmann K, Tan AH, Tay YW, Lim JL, et al. Clinical phenotype of Parkinson’s disease with a homozygous PRKN p.Cys441Arg mutation. Neurology Asia 2021;26:161–166.
- 49. Lee LV, Pascasio FM, Fuentes FD, Viterbo GH. Torsion dystonia in Panay, Philippines. Adv Neurol 1976;14:137–151.PubMed
- 50. Lee LV, Rivera C, Teleg RA, Dantes MB, Pasco PM, Jamora RD, et al. The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”). Int J Neurosci 2011;121 Suppl 1:3–11.ArticlePubMed
- 51. Lee LV, Munoz EL, Tan KT, Reyes MT. Sex linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Mol Pathol 2001;54:362–368.PubMedPMC
- 52. Ng AR, Jamora RDG, Rosales RL. X-linked dystonia Parkinsonism: crossing a new threshold. J Neural Transm (Vienna) 2021;128:567–573.ArticlePubMedPDF
- 53. Evidente VG, Gwinn-Hardy K, Hardy J, Hernandez D, Singleton A. X-linked dystonia (“Lubag”) presenting predominantly with parkinsonism: a more benign phenotype? Mov Disord 2002;17:200–202.ArticlePubMedPDF
- 54. Jamora RDG, Suratos CTR, Bautista JEC, Ramiro GMI, Westenberger A, Klein C, et al. Neurocognitive profile of patients with X-linked dystonia-parkinsonism. J Neural Transm (Vienna) 2021;128:671–678.ArticlePubMedPDF
- 55. Pasco PM, Ison CV, Muňoz EL, Magpusao NS, Cheng AE, Tan KT, et al. Understanding XDP through imaging, pathology, and genetics. Int J Neurosci 2011;121 Suppl 1:12–17.ArticlePubMed
- 56. Brüggemann N, Heldmann M, Klein C, Domingo A, Rasche D, Tronnier V, et al. Neuroanatomical changes extend beyond striatal atrophy in X-linked dystonia parkinsonism. Parkinsonism Relat Disord 2016;31:91–97.ArticlePubMed
- 57. Németh AH, Nolte D, Dunne E, Niemann S, Kostrzewa M, Peters U, et al. Refined linkage disequilibrium and physical mapping of the gene locus for X-linked dystonia-parkinsonism (DYT3). Genomics 1999;60:320–329.ArticlePubMed
- 58. Domingo A, Westenberger A, Lee LV, Brænne I, Liu T, Vater I, et al. New insights into the genetics of X-linked dystonia-parkinsonism (XDP, DYT3). Eur J Hum Genet 2015;23:1334–1340.ArticlePubMedPMCPDF
- 59. Nolte D, Niemann S, Müller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A 2003;100:10347–10352.ArticlePubMedPMC
- 60. Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet 2007;80:393–406.ArticlePubMedPMC
- 61. Westenberger A, Reyes CJ, Saranza G, Dobricic V, Hanssen H, Domingo A, et al. A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann Neurol 2019;85:812–822.ArticlePubMedPDF
- 62. Bragg DC, Sharma N, Ozelius LJ. X-Linked Dystonia-Parkinsonism: recent advances. Curr Opin Neurol 2019;32:604–609.ArticlePubMedPMC
- 63. Jamora RD, Diesta CC, Pasco PM, Lee LV. Oral pharmacological treatment of X-linked dystonia parkinsonism: successes and failures. Int J Neurosci 2011;121 Suppl 1:18–21.ArticlePubMed
- 64. Rosales RL, Santos MM, Ng AR, Teleg R, Dantes M, Lee LV, et al. The broadening application of chemodenervation in X-linked dystoniaparkinsonism (Part I): muscle afferent block versus botulinum toxin-A in cervical and limb dystonias. Int J Neurosci 2011;121 Suppl 1:35–43.ArticlePubMed
- 65. Kilbane C, Witt J, Galifianakis NB, Glass GA, Volz M, Heath S, et al. Long-term outcomes of bilateral pallidal deep brain stimulation for X-linked dystonia and parkinsonism. Stereotact Funct Neurosurg 2018;96:320–326.ArticlePubMedPDF
- 66. Brüggemann N, Domingo A, Rasche D, Moll CKE, Rosales RL, Jamora RDG, et al. Association of pallidal neurostimulation and outcome predictors with X-linked dystonia parkinsonism. JAMA Neurol 2019;76:211–216.ArticlePubMedPMC
- 67. Oyama G, Fernandez HH, Foote KD, Zeilman P, Hwynn N, Jacobson CE 4th, et al. Differential response of dystonia and parkinsonism following globus pallidus internus deep brain stimulation in X-linked dystonia-parkinsonism (Lubag). Stereotact Funct Neurosurg 2010;88:329–333.ArticlePubMedPMCPDF
- 68. Evidente VG, Lyons MK, Wheeler M, Hillman R, Helepolelei L, Beynen F, et al. First case of X-linked dystonia-parkinsonism (“Lubag”) to demonstrate a response to bilateral pallidal stimulation. Mov Disord 2007;22:1790–1793.ArticlePubMed
- 69. Wadia PM, Lim SY, Lozano AM, Adams JR, Poon YY, Torres Diaz CV, et al. Bilateral pallidal stimulation for x-linked dystonia parkinsonism. Arch Neurol 2010;67:1012–1015.ArticlePubMed
- 70. Smith JK, Gonda VE, Malamud N. Unusual form of cerebellar ataxia; combined dentato-rubral and pallido-Luysian degeneration. Neurology 1958;8:205–209.ArticlePubMed
- 71. Naito H, Izawa K, Kurosaki T, Kaji S, Sawa M. [Two families of progressive myoclonus epilepsy with Mendelian dominant heredity]. Seishin Shinkeigaku Zasshi 1972;74:871–897.Japanese. PubMed
- 72. Takahata N, Ito K, Yoshimura Y, Nishihori K, Suzuki H. Familial chorea and myoclonus epilepsy. Neurology 1978;28(9 Pt 1):913–919.ArticlePubMed
- 73. Naito H, Oyanagi S. Familial myoclonus epilepsy and choreoathetosis: hereditary dentatorubral-pallidoluysian atrophy. Neurology 1982;32:798–807.ArticlePubMed
- 74. Iizuka R, Hirayama K, Maehara KA. Dentato-rubro-pallido-luysian atrophy: a clinico-pathological study. J Neurol Neurosurg Psychiatry 1984;47:1288–1298.ArticlePubMedPMC
- 75. Takahashi H, Ohama E, Naito H, Takeda S, Nakashima S, Makifuchi T, et al. Hereditary dentatorubral-pallidoluysian atrophy: clinical and pathologic variants in a family. Neurology 1988;38:1065–1070.ArticlePubMed
- 76. Chaudhry A, Anthanasiou-Fragkouli A, Houlden H. DRPLA: understanding the natural history and developing biomarkers to accelerate therapeutic trials in a globally rare repeat expansion disorder. J Neurol 2021;268:3031–3041.ArticlePubMedPMCPDF
- 77. Farmer TW, Wingfield MS, Lynch SA, Vogel FS, Hulette C, Katchinoff B, et al. Ataxia, chorea, seizures, and dementia. Pathologic features of a newly defined familial disorder. Arch Neurol 1989;46:774–779.ArticlePubMed
- 78. Burke JR, Wingfield MS, Lewis KE, Roses AD, Lee JE, Hulette C, et al. The Haw River Syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African-American family. Nat Genet 1994;7:521–524.ArticlePubMedPDF
- 79. Kondo I, Ohta H, Yazaki M, Ikeda JE, Gusella JF, Kanazawa I. Exclusion mapping of the hereditary dentatorubropallidoluysian atrophy gene from the Huntington’s disease locus. J Med Genet 1990;27:105–108.ArticlePubMedPMC
- 80. Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993;4:221–226.ArticlePubMedPDF
- 81. Li SH, McInnis MG, Margolis RL, Antonarakis SE, Ross CA. Novel triplet repeat containing genes in human brain: cloning, expression, and length polymorphisms. Genomics 1993;16:572–579.ArticlePubMed
- 82. Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 1994;6:9–13.ArticlePubMedPDF
- 83. Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet 1994;6:14–18.ArticlePubMedPDF
- 84. Sato T, Oyake M, Nakamura K, Nakao K, Fukusima Y, Onodera O, et al. Transgenic mice harboring a full-length human mutant DRPLA gene exhibit age-dependent intergenerational and somatic instabilities of CAG repeats comparable with those in DRPLA patients. Hum Mol Genet 1999;8:99–106.ArticlePubMed
- 85. Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet 1983;20:216–219.ArticlePubMedPMC
- 86. Al-Awadi SA, Farag TI, Teebi AS, Naguib K, el-Khalifa MY, Kelani Y, et al. Primary hypogonadism and partial alopecia in three sibs with Müllerian hypoplasia in the affected females. Am J Med Genet 1985;22:619–622.ArticlePubMed
- 87. Gül D, Özata M, Mergen H, Odabasi Z, Mergen M. Woodhouse and Sakati syndrome (MIM 241080): report of a new patient. Clin Dysmorphol 2000;9:123–125.ArticlePubMed
- 88. Al-Semari A, Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am J Med Genet A 2007;143A:149–160.ArticlePubMed
- 89. Crandall BF, Samec L, Sparkes RS, Wright SW. A familial syndrome of deafness, alopecia, and hypogonadism. J Pediatr 1973;82:461–465.ArticlePubMed
- 90. Steindl K, Alazami AM, Bhatia KP, Wuerfel JT, Petersen D, Cartolari R, et al. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin Genet 2010;78:594–597.ArticlePubMed
- 91. Koshy G, Danda S, Thomas N, Mathews V, Viswanathan V. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin Dysmorphol 2008;17:57–60.ArticlePubMed
- 92. Habib R, Basit S, Khan S, Khan MN, Ahmad W. A novel splice site mutation in gene C2orf37 underlying Woodhouse-Sakati syndrome (WSS) in a consanguineous family of Pakistani origin. Gene 2011;490(1-2):26–31.ArticlePubMed
- 93. Hdiji O, Turki E, Bouzidi N, Bouchhima I, Damak M, Bohlega S, et al. Woodhouse-Sakati syndrome: report of the first Tunisian family with the C2orf37 gene mutation. J Mov Disord 2016;9:120–123.PubMedPMC
- 94. Louro P, Durães J, Oliveira D, Paiva S, Ramos L, Macário MC. Woodhouse-Sakati syndrome: first report of a Portuguese case. Am J Med Genet A 2019;179:2237–2240.ArticlePubMedPDF
- 95. Ali R, Al-Dewik N, Mohammed S, Elfituri M, Agouba S, Musa S, et al. Expanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17: report of 58 additional patients from Qatar and literature review. Am J Med Genet A 2022;188:116–129.PubMed
- 96. Schneider SA, Bhatia KP. Dystonia in the Woodhouse Sakati syndrome: a new family and literature review. Mov Disord 2008;23:592–596.ArticlePubMed
- 97. Bohlega S, Abusrair AH, Al-Ajlan FS, Alharbi N, Al-Semari A, Bohlega B, et al. Patterns of neurological manifestations in Woodhouse-Sakati syndrome. Parkinsonism Relat Disord 2019;69:99–103.ArticlePubMed
- 98. Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet 2008;83:684–691.ArticlePubMedPMC
- 99. Ben-Omran T, Ali R, Almureikhi M, Alameer S, Al-Saffar M, Walsh CA, et al. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: two new families with a mutation in the C2orf37 gene. Am J Med Genet A 2011;155A:2647–2653.ArticlePubMedPMC
- 100. Abusrair AH, Bohlega S, Al-Semari A, Al-Ajlan FS, Al-Ahmadi K, Mohamed B, et al. Brain MR imaging findings in Woodhouse-Sakati syndrome. AJNR Am J Neuroradiol 2018;39:2256–2262.ArticlePubMedPMC
- 101. Almeqdadi M, Kemppainen JL, Pichurin PN, Gavrilova RH. Phenotypic variability of c.436delC DCAF17 gene mutation in Woodhouse-Sakati syndrome. Am J Case Rep 2018;19:347–353.ArticlePubMedPMC
- 102. Bohlega SA, Abusrair A. Woodhouse-Sakati syndrome [updated 2021 Jul 8]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al. GeneReviews(R) [Internet]. Seattle: University of Washington. c1993-2023 [accessed on 2022 Mar 13]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK378974/.
- 103. Wakeno M. [A family with heredofamilial tremor associated with epileptic disorders]. Seishin Shinkeigaku Zasshi 1975;77:1–18.Japanese. PubMed
- 104. Kudo J, Kudo T, Yamauchi T. [Seven families with heredofamilial tremor and epilepsy]. Rinsho Shinkeigaku 1984;24:1–8.Japanese. PubMed
- 105. Inoue S. [On a pedigree of the hereditary tremor with epileptiform seizures]. Psychiat Neurol Jpn 1951;53:33–37.Japanese.
- 106. Inazuki G, Naito H, Ohama E, Kawase Y, Honma Y, Tokiguchi S, et al. [A clinical study and neuropathological findings of a familial disease with myoclonus and epilepsy--the nosological place of familial essential myoclonus and epilepsy (FEME)]. Seishin Shinkeigaku Zasshi 1990;92:1–21.Japanese. PubMed
- 107. Nakazawa F. [A hereditary neurological disease presenting a kind of tremor and epileptic attacks resembling Unverricht-Lundborg’s myoclonus epilepsy]. Psychiat Neurol Jpn 1924;24:484–495.Japanese.
- 108. Naito H, Kaji S. [Clinical picture and type of myoclonus epilepsy with dominant heredity (author’s transl)]. Seishin Shinkeigaku Zasshi 1979;81:571–586.Japanese. PubMed
- 109. Ikeda A, Kakigi R, Funai N, Neshige R, Kuroda Y, Shibasaki H. Cortical tremor: a variant of cortical reflex myoclonus. Neurology 1990;40:1561–1565.ArticlePubMed
- 110. Yasuda T. Benign adult familial myoclonic epilepsy (BAFME). Kawasaki Med J 1991;17(1-4):1–13.
- 111. Kobayashi K, Hitomi T, Matsumoto R, Watanabe M, Takahashi R, Ikeda A. Nationwide survey in Japan endorsed diagnostic criteria of benign adult familial myoclonus epilepsy. Seizure 2018;61:14–22.ArticlePubMed
- 112. Hitomi T, Kobayashi K, Sakurai T, Ueda S, Jingami N, Kanazawa K, et al. Benign adult familial myoclonus epilepsy is a progressive disorder: no longer idiopathic generalized epilepsy. Epileptic Disord 2016;18:67–72.ArticlePubMed
- 113. Neshige S, Hitomi T, Tojima M, Oi K, Kobayashi K, Matsuhashi M, et al. A role of aging in the progression of cortical excitability in benign adult familial myoclonus epilepsy type 1 patients. Mov Disord 2021;36:2446–2448.ArticlePubMedPDF
- 114. Plaster NM, Uyama E, Uchino M, Ikeda T, Flanigan KM, Kondo I, et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology 1999;53:1180–1183.ArticlePubMed
- 115. Mikami M, Yasuda T, Terao A, Nakamura M, Ueno S, Tanabe H, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am J Hum Genet 1999;65:745–751.ArticlePubMedPMC
- 116. Suzuki T. [Clinical genetics and linkage analysis of familial essential myoclonus and epilepsy (FEME)]. Niigata Igakkai Zasshi 2002;116:535–545.Japanese.
- 117. Mori S, Nakamura M, Yasuda T, Ueno S, Kaneko S, Sano A. Remapping and mutation analysis of benign adult familial myoclonic epilepsy in a Japanese pedigree. J Hum Genet 2011;56:742–747.ArticlePubMedPDF
- 118. Cen ZD, Xie F, Lou DN, Ouyang ZY, Liu L, Cao J, et al. Fine mapping and whole-exome sequencing of a familial cortical myoclonic tremor with epilepsy family. Am J Med Genet B Neuropsychiatr Genet 2015;168:595–599.ArticlePubMed
- 119. Guerrini R, Bonanni P, Patrignani A, Brown P, Parmeggiani L, Grosse P, et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: A newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain 2001;124(Pt 12):2459–2475.ArticlePubMed
- 120. de Falco FA, Striano P, de Falco A, Striano S, Santangelo R, Perretti A, et al. Benign adult familial myoclonic epilepsy: genetic heterogeneity and allelism with ADCME. Neurology 2003;60:1381–1385.ArticlePubMed
- 121. Striano P, Madia F, Minetti C, Striano S, Zara F. Electroclinical and genetic findings in a family with cortical tremor, myoclonus, and epilepsy. Epilepsia 2005;46:1993–1995.ArticlePubMed
- 122. Suppa A, Berardelli A, Brancati F, Marianetti M, Barrano G, Mina C, et al. Clinical, neuropsychological, neurophysiologic, and genetic features of a new Italian pedigree with familial cortical myoclonic tremor with epilepsy. Epilepsia 2009;50:1284–1288.ArticlePubMed
- 123. Crompton DE, Sadleir LG, Bromhead CJ, Bahlo M, Bellows ST, Arsov T, et al. Familial adult myoclonic epilepsy: recognition of mild phenotypes and refinement of the 2q locus. Arch Neurol 2012;69:474–481.ArticlePubMed
- 124. Henden L, Freytag S, Afawi Z, Baldassari S, Berkovic SF, Bisulli F, et al. Identity by descent fine mapping of familial adult myoclonus epilepsy (FAME) to 2p11.2-2q11.2. Hum Genet 2016;135:1117–1125.ArticlePubMedPDF
- 125. Depienne C, Magnin E, Bouteiller D, Stevanin G, Saint-Martin C, Vidailhet M, et al. Familial cortical myoclonic tremor with epilepsy: the third locus (FCMTE3) maps to 5p. Neurology 2010;74:2000–2003.ArticlePubMed
- 126. Yeetong P, Ausavarat S, Bhidayasiri R, Piravej K, Pasutharnchat N, Desudchit T, et al. A newly identified locus for benign adult familial myoclonic epilepsy on chromosome 3q26.32-3q28. Eur J Hum Genet 2013;21:225–228.ArticlePubMedPMCPDF
- 127. Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet 2018;50:581–590.PubMed
- 128. Hitomi T, Kobayashi K, Jingami N, Nakagawa T, Imamura H, Matsumoto R, et al. Increased clinical anticipation with maternal transmission in benign adult familial myoclonus epilepsy in Japan. Epileptic Disord 2013;15:428–432.ArticlePubMed
- 129. Bennett MF, Oliver KL, Regan BM, Bellows ST, Schneider AL, Rafehi H, et al. Familial adult myoclonic epilepsy type 1 SAMD12 TTTCA repeat expansion arose 17,000 years ago and is present in Sri Lankan and Indian families. Eur J Hum Genet 2020;28:973–978.ArticlePubMedPMCPDF
- 130. Mahadevan R, Bhoyar RC, Viswanathan N, Rajagopal RE, Essaki B, Suroliya V, et al. Genomic analysis of patients in a South Indian community with autosomal dominant cortical tremor, myoclonus and epilepsy suggests a founder repeat expansion mutation in the SAMD12 gene. Brain Commun 2021;3:fcaa214.ArticlePubMedPMCPDF
- 131. Yeetong P, Chunharas C, Pongpanich M, Bennett MF, Srichomthong C, Pasutharnchat N, et al. Founder effect of the TTTCA repeat insertions in SAMD12 causing BAFME1. Eur J Hum Genet 2021;29:343–348.ArticlePubMedPMCPDF
- 132. Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain 2018;141:2280–2288.ArticlePubMed
- 133. Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun 2019;10:4920.ArticlePubMedPMCPDF
- 134. Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, et al. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 2019;142:3360–3366.ArticlePubMedPDF
- 135. Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic Epilepsy type 3. Nat Commun 2019;10:4919.PubMedPMC
- 136. Ishiura H, Tsuji S. Advances in repeat expansion diseases and a new concept of repeat motif-phenotype correlation. Curr Opin Genet Dev 2020;65:176–185.ArticlePubMed
- 137. Oi K, Neshige S, Hitomi T, Kobayashi K, Tojima M, Matsuhashi M, et al. Low-dose perampanel improves refractory cortical myoclonus by the dispersed and suppressed paroxysmal depolarization shifts in the sensorimotor cortex. Clin Neurophysiol 2019;130:1804–1812.ArticlePubMed
- 138. Najim Al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 1994;89:347–352.ArticlePubMed
- 139. Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ. Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 2005;20:1264–1271.ArticlePubMed
- 140. Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wriekat AL, et al. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet 2001;38:680–682.ArticlePubMedPMC
- 141. Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006;38:1184–1191.ArticlePubMedPDF
- 142. Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 2010;25:1929–1937.ArticlePubMed
- 143. Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007;68:1557–1562.ArticlePubMed
- 144. De Michele G, Galatolo D, Lieto M, Fico T, Saccà F, Santorelli FM, et al. Ataxia-myoclonus syndrome due to a novel homozygous ATP13A2 mutation. Parkinsonism Relat Disord 2020;76:42–43.ArticlePubMed
- 145. Rohani M, Lang AE, Sina F, Elahi E, Fasano A, Hardy J, et al. Action myoclonus and seizure in Kufor-Rakeb syndrome. Mov Disord Clin Pract 2017;5:195–199.ArticlePubMedPMCPDF
- 146. Pietrzak A, Badura-Stronka M, Kangas-Kontio T, Felczak P, Kozubski W, Latos-Bielenska A, et al. Clinical and ultrastructural findings in an ataxic variant of Kufor-Rakeb syndrome. Folia Neuropathol 2019;57:285–294.ArticlePubMed
- 147. Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet 2012;21:2646–2650.ArticlePubMedPMC
- 148. Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T, et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017;140:287–305.ArticlePubMedPMC
- 149. Spataro R, Kousi M, Farhan SMK, Willer JR, Ross JP, Dion PA, et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum Genomics 2019;13:19.ArticlePubMedPMCPDF
- 150. Balint B, Damasio J, Magrinelli F, Guerreiro R, Bras J, Bhatia KP. Psychiatric manifestations of ATP13A2 mutations. Mov Disord Clin Pract 2020;7:838–841.ArticlePubMedPMCPDF
- 151. Kruer MC, Paudel R, Wagoner W, Sanford L, Kara E, Gregory A, et al. Analysis of ATP13A2 in large neurodegeneration with brain iron accumulation (NBIA) and dystonia-parkinsonism cohorts. Neurosci Lett 2012;523:35–38.ArticlePubMedPMC
- 152. Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep 2011;11:254–261.ArticlePubMedPMCPDF
- 153. Bohlega S, Stigsby B, Al-Kawi MZ, McLean DR, Ozand P, Omer S, et al. Familial tremulous and myoclonic dystonia with white matter changes in brain magnetic resonance imaging. Mov Disord 1995;10:513–517.ArticlePubMed
- 154. Monies D, Abou Al-Shaar H, Goljan EA, Al-Younes B, Al-Breacan MMA, Al-Saif MM, et al. Identification of a novel genetic locus underlying tremor and dystonia. Hum Genomics 2017;11:25.ArticlePubMedPMCPDF
- 155. Mount LA, Reback S. Familial paroxysmal choreoathetosis: preliminary report on a hitherto undescribed clinical syndrome. Arch Neur Psych 1940;44:841–847.
- 156. Kertesz A. Paroxysmal kinesigenic choreoathetosis. An entity within the paroxysmal choreoathetosis syndrome. Description of 10 cases, including 1 autopsied. Neurology 1967;17:680–690.ArticlePubMed
- 157. Kato N, Sadamatsu M, Kikuchi T, Niikawa N, Fukuyama Y. Paroxysmal kinesigenic choreoathetosis: from first discovery in 1892 to genetic linkage with benign familial infantile convulsions. Epilepsy Res 2006;70 Suppl 1:S174–S184.ArticlePubMed
- 158. Kure S. Atypical Thomsen’s disease. Tokyo Igakukai Zasshi (J Tokyo Med Assoc) 1892;6:505–514.
- 159. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain 2015;138(Pt 12):3567–3580.ArticlePubMedPMC
- 160. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol 1995;38:571–579.ArticlePubMed
- 161. Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet 2011;43:1252–1255.ArticlePubMedPDF
- 162. Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang JG, et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011;134(Pt 12):3493–3501.ArticlePubMedPMC
- 163. Lee HY, Xu Y, Huang Y, Ahn AH, Auburger GW, Pandolfo M, et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet 2004;13:3161–3170.ArticlePubMed
- 164. Rainier S, Thomas D, Tokarz D, Ming L, Bui M, Plein E, et al. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol 2004;61:1025–1029.ArticlePubMed
- 165. ang D, Kranz-Eble P, De Vivo DC. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum Mutat 2000;16:224–231.ArticlePubMed
- 166. Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord 2014;29:1108–1116.ArticlePubMedPDF
- 167. Jagota P, Lim SY, Pal PK, Lee JY, Kukkle PL, Fujioka S, et al. Genetic movement disorders commonly seen in Asians. Mov Disord Clin Prac 2023;10:878–895.Article
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite