E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 10(1); 2017 > Article
-
Case Report
Suspected Perinatal Depression Revealed to be Hereditary Diffuse Leukoencephalopathy with Spheroids - Josefine Blume, Robert Weissert
-
Journal of Movement Disorders 2017;10(1):59-61.
DOI: https://doi.org/10.14802/jmd.16050
Published online: December 27, 2016
Department of Neurology, University of Regensburg, Regensburg, Germany
- Corresponding author: Josefine Blume, MD, PhD, Department of Neurology, University of Regensburg, Universitaetsstrasse 84, Regensburg 93053, Germany / Tel: +49-0941-9410 / Fax: +49-0941-9413105 / E-mail: joblume.rb@gmail.com
• Received: October 10, 2016 • Revised: November 1, 2016 • Accepted: November 18, 2016
Copyright © 2017 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Early motor symptoms of neurodegenerative diseases often appear in combination with psychiatric symptoms, such as depression or personality changes, and are in danger of being misdiagnosed as psychogenic in young patients. We present the case of a 32-year-old woman who presented with rapid-onset depression, followed by a hypokinetic movement disorder and cognitive decline during pregnancy. Genetic testing revealed a mutation in the colony-stimulating factor 1 receptor gene, which led to the diagnosis of hereditary diffuse leukoencephalopathy with spheroids. Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is probably an under-recognized disease. HDLS should be considered in patients with rapidly progressing parkinsonian symptoms and dementia accompanied by white matter lesions.
- A 32-year-old female patient developed depression, anxiety and subtle gait disturbances during the second trimester of her first, otherwise uncomplicated pregnancy. Her main complaint upon her first visit to the emergency department was having difficulty finding words and concentrating, as well as having a fear of falling while walking. She felt anxious and hopeless and showed mildly decreased cognitive function, achieving a score of 24 points on the Mini Mental State Examination. Perinatal depression was suspected, and she was treated by the Department of Psychiatry for six months without significant improvement in her symptoms. Upon completion of her treatment, the patient displayed an unusual wide-based, shuffling, very slow and highly fluctuating gait. She walked in small steps and sometimes staggered severely, but her symptoms were variable, and she did not fall. Therefore, her gait disturbances were classified as psychogenic. However, cerebral MRI showed confluent white matter lesions suspicious for CADASIL. The patient had been treated for mild hypertension since the age of 29 but had otherwise been healthy. Her family history was negative for any hereditary diseases, but her reported history was fragmented because she had broken off all contact with her father at the age of 18.
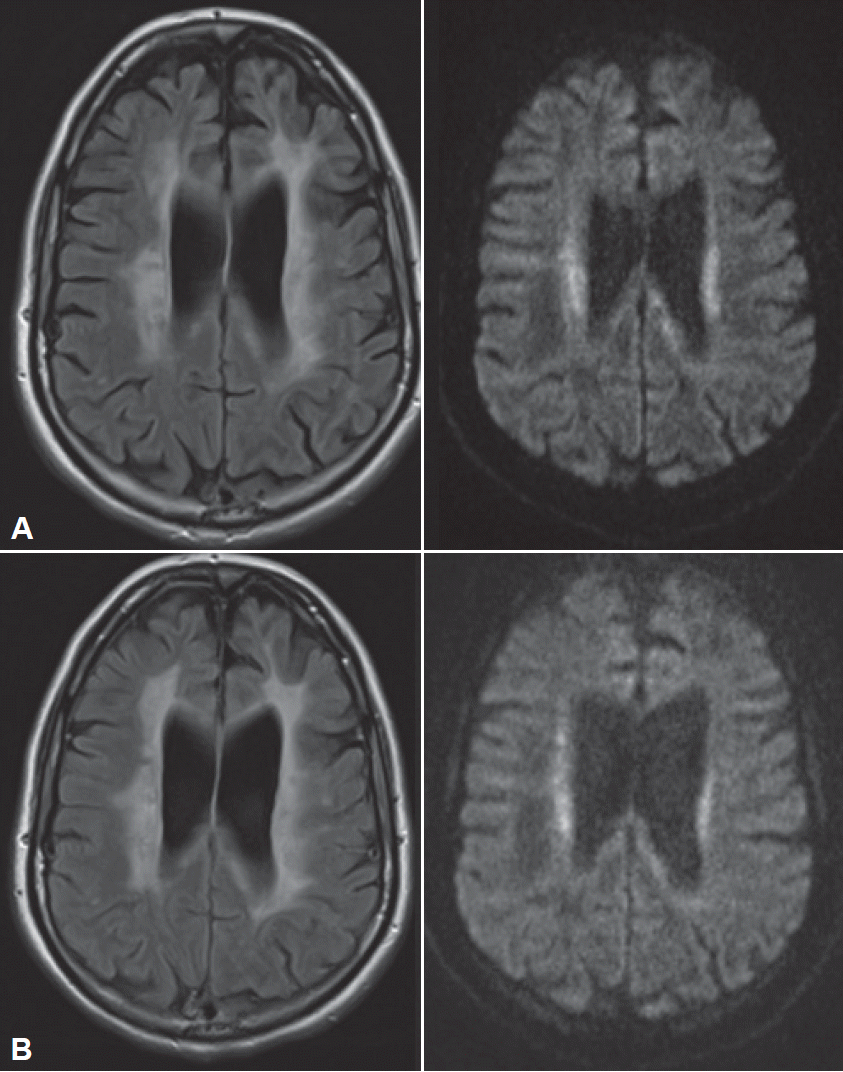
- The patient subsequently presented to us ten months after symptom onset and six months after she had given birth to a healthy girl. She presented with conspicuous global bradykinesia with severe slowing and hesitation in her fine motor skills and symmetric rigidity in all her extremities, but without tremor. She also exhibited reduced spontaneous speech with slight amnestic aphasia and ataxic dysarthria, with loss of modulation. Her gait disturbances had worsened, as she could walk only short distances independently, and she had difficulty lifting her feet of the ground without external instruction but showed no typical freezing behaviors. Apraxia was an important finding, as it was evident in both her fine motor skills and her gait. Another MRI revealed the presence of increasingly symmetrical, confluent FLAIR hyperintensities with partly restricted diffusion, but without contrast enhancement (Figure 1). Wideranging blood and CSF analyses, as well as electrophysiological tests, were not suggestive of a diagnosis. In particular, there was no evidence of an infectious or autoimmune cause of her symptoms.
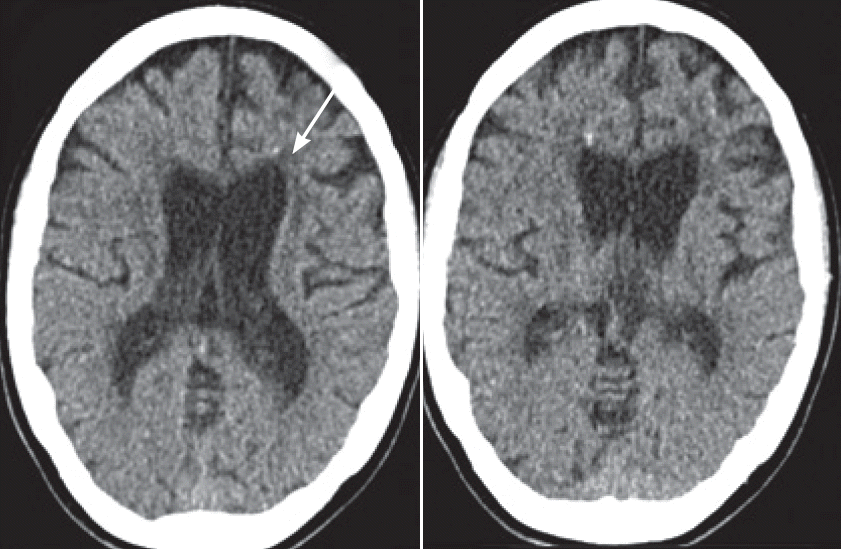
- The marked parkinsonian features, which improved slightly on levodopa, combined with the progressive leukoencephalopathy and spotty frontal calcifications demonstrated by CT (Figure 2) led us to test for HDLS. Genetic testing revealed the presence of a heterozygous mutation (c.2541G>C) in the CSF1R gene leading to a change in the corresponding amino acid sequence (p.E847D). This mutation was first described in a patient who presented with cognitive decline and spastic paraparesis at the age of 44 [3].
- The patient exhibited signs of progressive pyramidal as well as extrapyramidal motor dysfunction and rapidly progressing dementia during the following months (Supplementary Video 1 in the online-only Data Supplement). Eighteen months after symptom onset, the patient was admitted to a nursing home. By that time, she was not able to sit, stand, communicate or recognize faces. She presented with a combination of rigid-spastic muscle tonus, pyramidal signs and primitive reflexes. The patient died 28 months after symptom onset.
CASE REPORT
- This young woman’s case was highly suspicious for infection or autoimmune disease due to its subacute onset and rapid progression during her pregnancy and shortly after her first childbirth. HDLS is caused by mutations in the CSF1R gene. CSF1R and its ligands, CSF1 and IL-34, are required for placental development [9]. We therefore hypothesize that the extensive adaptations of the maternal immune system that occur during pregnancy contribute to the clinical manifestations of the disease. Further research is needed to prove this theory.
- In addition to the parkinsonian features, the spotty calcifications that were noted in the affected frontal white matter on CT were a hint to the diagnosis. These findings were first described by Fujioka et al. [10], who reported the case of a female patient with a CSF1R mutation in 2013, and Konno et al. [8], who presented the results pertaining to a set of patients in 2014. CSF1R signaling is known to be necessary for osteoclast cytoskeletal reorganization. Therefore, a direct pathogenic relationship between CSF1R signaling and calcification is conceivable. The calcifications seem to be specific for HDLS, but this specificity is not yet common knowledge. CT should be performed in suspected cases to confirm the diagnosis and to investigate the specificity of this finding further.
- Early motor symptoms of neurodegenerative diseases, which often appear in combination with psychiatric symptoms, such as depression or personality changes, are in danger of being misdiagnosed as psychogenic in young patients, especially during and shortly after pregnancy. HDLS is probably an underrecognized disease. HDLS should be considered in patients with rapidly progressing parkinsonian symptoms and dementia accompanied by white matter lesions.
DISCUSSION
Supplementary Materials
Supplementary Video Legend
Figure 1.Cerebral MRI at the first visit (A) and nine months later (B). A: Cerebral MRI at the first visit. Confluent hyperintensities in the periventricular and deep white matter (FLAIR, left) with partly restricted diffusion (diffusion-weighted, right). B: Cerebral MRI nine months later: increasing hyperintensities affecting almost the entire white matter (FLAIR, left). FLAIR: fluid-attenuated inversion recovery.
Figure 2.Cerebral CT at the first visit. Generalized supratentorial atrophy inconsistent with an age of 32 years and multiple spotty calcifications in the frontal white matter (arrow).
- 1. Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 2016;87:512–519.ArticlePubMedPMC
- 2. Hoffmann S, Murrell J, Harms L, Miller K, Meisel A, Brosch T, et al. Enlarging the nosological spectrum of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Brain Pathol 2014;24:452–458.ArticlePubMedPMC
- 3. Guerreiro R, Kara E, Le Ber I, Bras J, Rohrer JD, Taipa R, et al. Genetic analysis of inherited leukodystrophies: genotype- phenotype correlations in the CSF1R gene. JAMA Neurol 2013;70:875–882.ArticlePubMedPMC
- 4. Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2011;44:200–205.ArticlePubMedPMCPDF
- 5. Chitu V, Gokhan S, Gulinello M, Branch CA, Patil M, Basu R, et al. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol Dis 2015;74:219–228.ArticlePubMedPMC
- 6. Chitu V, Gokhan Ş, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci 2016;39:378–393.ArticlePubMedPMC
- 7. Sundal C, Jönsson L, Ljungberg M, Zhong J, Tian W, Zhu T, et al. Different stages of white matter changes in the original HDLS family revealed by advanced MRI techniques. J Neuroimaging 2014;24:444–452.ArticlePubMed
- 8. Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, et al. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2014;82:139–148.ArticlePubMedPMC
- 9. Pampfer S, Daiter E, Barad D, Pollard JW. Expression of the colony-stimulating factor-1 receptor (c-fms proto-oncogene product) in the human uterus and placenta. Biol Reprod 1992;46:48–57.ArticlePubMed
- 10. Fujioka S, Broderick DF, Sundal C, Baker MC, Rademakers R, Wszolek ZK. An adult-onset leukoencephalopathy with axonal spheroids and pigmented glia accompanied by brain calcifications: a case report and a literature review of brain calcifications disorders. J Neurol 2013;260:2665–2668.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Modeling CSF‐1 receptor deficiency diseases – how close are we?
Violeta Chitu, Şölen Gökhan, E. Richard Stanley
The FEBS Journal.2022; 289(17): 5049. CrossRef - Neuroimaging phenotypes of CSF1R‐related leukoencephalopathy: Systematic review, meta‐analysis, and imaging recommendations
Goda‐Camille Mickeviciute, Monika Valiuskyte, Michael Plattén, Zbigniew K. Wszolek, Oluf Andersen, Virginija Danylaité Karrenbauer, Benjamin V. Ineichen, Tobias Granberg
Journal of Internal Medicine.2022; 291(3): 269. CrossRef - A Novel Missense Mutation of the CSF1R Gene Causes Incurable CSF1R-Related Leukoencephalopathy: Case Report and Review of Literature
Jie Chen, Shiying Luo, Ning Li, Huimin Li, Jinming Han, Li Ling
International Journal of General Medicine.2020; Volume 13: 1613. CrossRef -
CSF1R
-related leukoencephalopathy
Takuya Konno, Koji Kasanuki, Takeshi Ikeuchi, Dennis W. Dickson, Zbigniew K. Wszolek
Neurology.2018; 91(24): 1092. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite