E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 10(1); 2017 > Article
-
Case Report
Familial Hyperekplexia, a Potential Cause of Cautious Gait: A New Korean Case and a Systematic Review of Phenotypes - Yoonju Lee1, Nan Young Kim2, Sangkyoon Hong2, Su Jin Chung1, Seong Ho Jeong1, Phil Hyu Lee1, Young H. Sohn1
-
Journal of Movement Disorders 2017;10(1):53-58.
DOI: https://doi.org/10.14802/jmd.16044
Published online: December 27, 2016
1Department of Neurology, Yonsei University College of Medicine, Seoul, Korea
2Hallym Institute of Translational Genomics and Bioinformatics, Hallym University College of Medicine, Anyang, Korea
- Corresponding author: Young H. Sohn, MD, PhD, Department of Neurology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea / Tel: +82-2-2228-1601 / Fax: +82-2-393-0705 / E-mail: yhsohn62@yuhs.ac
Copyright © 2017 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Familial hyperekplexia, also called startle disease, is a rare neurological disorder characterized by excessive startle responses to noise or touch. It can be associated with serious injury from frequent falls, apnea spells, and aspiration pneumonia. Familial hyperekplexia has a heterogeneous genetic background with several identified causative genes; it demonstrates both dominant and recessive inheritance in the α1 subunit of the glycine receptor (GLRA1), the β subunit of the glycine receptor and the presynaptic sodium and chloride-dependent glycine transporter 2 genes. Clonazepam is an effective medical treatment for hyperekplexia. Here, we report genetically confirmed familial hyperekplexia patients presenting early adult cautious gait. Additionally, we review clinical features, mode of inheritance, ethnicity and the types and locations of mutations of previously reported hyperekplexia cases with a GLRA1 gene mutation.
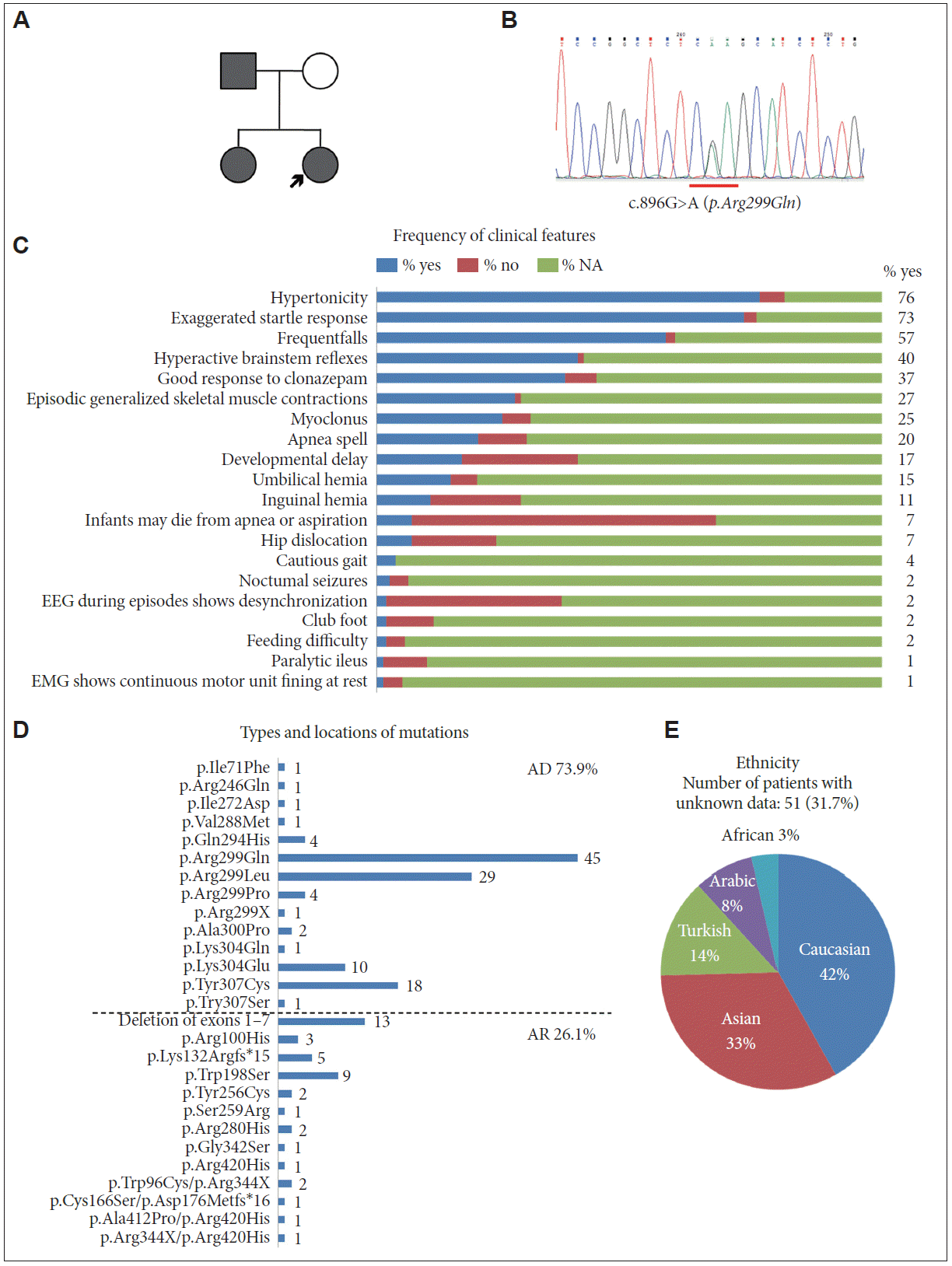
- A 20-year-old woman visited the neurology clinic for generalized stiffness and frequent falling episodes secondary to tactile stimuli. She was born at term, and her antenatal and birth history were not remarkable. There was no developmental delay or neurologic deficit; however, her parents had noticed sudden falling events since she was five years old. In response to unexpected tactile stimulation, she felt her body become rigid for a few seconds, which resulted in injurious falling down events with spared consciousness. She usually kept indoors and walked cautiously in order to avoid unexpected falling accidents. In childhood, the frequency of her falls was approximately four or five times per year, but after her teenage years, the frequency decreased to once or twice per year. She remembered that drinking alcohol ameliorated the symptoms. Her father and older sister had similar symptoms (Figure 1A). On physical examination, she had numerous scars on her forehead from previous falling accidents. Apart from cautious gait, her neurological examination was normal. Brain MRI and EEG were not remarkable.
- Whole exome sequencing with genomic DNA extracted from peripheral blood identified a heterozygous missense mutation c.896G>A (reference sequence: NM_001146040.1) in GLRA1. No mutations were found in other genes known to cause familial hyperekplexia, such as GLRB, SLC6A5, GPHN, and ARHGEF9. The change in the patient’s GLRA1 sequence alters the arginine codon at 299 to a glycine codon (p.Arg299Gln). This mutation was confirmed by Sanger sequencing (Figure 1B). The same mutation was also found in her sister, who was symptomatic; however, we could not perform a genetic study on her parents. Clonazepam was administered at a dose of 0.5 mg per day, which resulted in an improvement in the startle response.
CASE REPORT
- Hyperekplexia, known as a hereditary startle disease, is characterized by an exaggerated startle response and neonatal hypertonia. This disorder is a rare neurogenetic condition, but it is potentially treatable [1]. The symptom spectrum can vary from an exaggerated startle response to infantile apnea spells and even injurious falls. The disease can be accompanied by abdominal hernia, hip dislocation and developmental delay [2]. A previous case study reported a possible association with sudden infant death syndrome [3]. In patients with hyperekplexia, no abnormalities are observed on routine blood tests, urinalysis, brain imaging studies, or EEG [1]. Hyperekplexia could be misdiagnosed as epilepsy, cerebral palsy, anxiety disorder, or conversion disorder and therefore can be mistreated. Early diagnosis and treatment are important, as they not only prevent injuries but may also influence the quality of life of a patient.
- To conduct a systematic review of the literature regarding hyperekplexia cases caused by mutation in the GLRA1 gene, we retrieved articles from the PubMed database using the keywords “Hyperekplexia AND GLRA1, English” and “Hyperekplexia AND case, English”. The references used in this systematic review are listed in the supplementary information. Clinical features, ethnicity, types and locations of mutations and mode of inheritance, as obtained from the retrieved literature, are summarized in Table 1. Most patients showed neonatal hypertonia (76%) and an exaggerated startle response (73%). Most patients (64 out of 66 cases with the nose-tapping test) exhibited a hyperactive brainstem reflex, which was found with the nose-tapping test. Exaggerated head retraction reflexes in response to the nose-tapping test indicate exaggerated brainstem reflexes and provide an important clue to diagnose hyperekplexia. The patients also suffered from severe complications, such as developmental delay (16.8%) and apnea spells (20.7%). External abnormalities, such as umbilical (13.9%) and inguinal hernia (11.2%), hip dislocation (6.8%) and club foot (1.9%), were not uncommonly observed (Figure 1C). These findings are consistent with a previous case series that is not included in our analysis [4]. Clinical features that helped differentiate hyperekplexia from epilepsy included unexpected stimulus-inducing falling accidents, short episodes lasting only a few seconds, and spared consciousness, with no other abnormal movements accompanying the event. Cautious gait, face lacerations and family history may be helpful for differentiating hyperekplexia from conversion disorder. Frequent falls were observed in 39.8% of cases for which information was available, and cautious gait was reported infrequently (4%). Data regarding falls and gait might have been biased by patients’ age, and therefore, there is a potential for missed information. Six out of 161 reviewed patients exhibited a wide-based and stiff gait due to considerable fear of an unexpected falling event, and two patients lacked confidence in outdoor environments, resulting in impaired social behavior. Presentation of a cautious gait resulting from unexpected falling episodes might be an indication of hyperekplexia. Clonazepam, which enhances GABA-gated chloride channel function and presumably compensates for defective glycine-gated chloride channel function, has been considered the first choice for the treatment of hyperekplexia. Antiepileptic drugs, including carbamazepine, phenytoin, valproate, and vigabatrin, have also been used for treatment [1]. In this review, 60 out of 70 cases (85.7%) showed good response to clonazepam, which is similar to a previous study [4].
- Among genes causing familial hyperekplexia, GLRA1 is the most common causative gene, accounting for 80% of hereditary cases [5,6]. Our patients carried a heterozygous mutation, p.Arg299Gln, which was inherited in an autosomal dominant fashion. Missense mutation of the arginine at codon 299, which was previously reported as codon 271, is the most common (Table 1, Figure 1D). Both autosomal dominant and recessive inheritance have been reported in familial hyperekplexia caused by GLRA1 mutation. Our analysis showed that dominant inheritance (73.9%) was 3-fold more commonly reported than recessive inheritance (26.1%). Interestingly, most dominantly inherited mutations were located between codons 290–300 of the GLRA1 gene (Figure 1D). Distribution of ethnicity in the reviewed hyperekplexia cases was Caucasian (42%), Asian (33%), Turkish (14%), Arabic (8%), and African (3%) (Figure 1E). In a genotype-ethnicity correlation, 8 Asian families (including isolated cases) and 7 Caucasian families demonstrated the p.Arg299Gln mutation of the GLRA1 gene. These findings support the notion that the Arg299 amino acid site is vulnerable to hyperekplexia in ethnically disparate cases [7].
DISCUSSION
Supplementary Materials

| Studies (years) | Study design | n cases | Male (%) | n of family and case | Mode of inheritance | Ethnicities | Age of onset | Symtpoms | Reported mutations | Mutation position according to NP_000162 |
|---|---|---|---|---|---|---|---|---|---|---|
| Present family | Family study | 3 | 33 | 1 | AD | Asian | C | SR (2), FI, NT | p.Arg271n | p.Arg299Gln |
| Mine et al. (2015) | Original article | 16* | 50 | 11 | AD (14), AR(2) | Asian | I | NH, SR, AS (few), DD (1), NT, UH (10) | p.Arg271Gln (10) | p.Arg299Gln |
| p.Ala272Pro (2) | p.Ala300Pro | |||||||||
| p.Tyr279Cys (1) | p.Tyr307Cys | |||||||||
| p.Lys276Glu (1) | p.Lys304Glu | |||||||||
| p.Ala384Pro/p.Arg392His (1) | p.Ala412Pro/p.Arg420His | |||||||||
| p.Arg316X/p.Arg392His (1) | p.Arg344X/p.Arg420His | |||||||||
| Hmami et al. (2014) | Case report/series | 1 | 100 | 1‡ | AR | African | I | NH, SR, AS, NT | p.Arg392His | p.Arg420His |
| Horváth et al. (2014) | Case report/series | 1 | 100 | 1 | AD | Asian | I | NH, HD | p.lle43e | p.lle71Phe |
| Lee et al. (2013) | Original article | 1* | 100 | 1 | AD | Asian | I | NH, SR, FD | p.Arg271 | p.Arg299X |
| Chan et al. (2014) | Case report/series | 1 | 100 | 1 | AR | Asian | I | SR, Rg, FI, DD, NT | p.Cys138Ser/p.Asp148Metfs*16 | p.Cys166Ser/p.Asp176Metfs*16 |
| Zoons et al. (2012) | Family study | 5 | 20 | 1 | AD | Caucasian | I (1), U (4) | NH (4), SR (4), FI (4), DM (4), NT | p.Lys104Argfs*15 | p.Lys132Argfs*15 |
| Al-Futaisi et al. (2012) | Family study | 9 | 78 | 2‡ | AR | Arabic | I (6), C (3) | NH (6), SR (9), AS (1), DD (8), NT (1) | p.Trp170Ser | p.Trp198Ser |
| Gregory et al. (2008) | Family study | 4 | 50 | 1‡ | AD | African | I (2), C (2) | NH (2), SR (3), AS (1), FD (1), DD (2), UH (2) | p.Arg271Pro | p.Arg299Pro |
| Kang et al. (2008) | Case report/series | 1 | 100 | 1 | AD | Asian | I | NH, SR, DM, NT | p.Lys276Gln | p.Lys304Gln |
| Forsyth et al. (2007) | Case report/series | 2 | 0 | 1‡ | AR | Turkish | I | NH (1), SR, Rg (1), FI (1), AS (1), DD, NT | p.Tyr228Cys | p.Tyr256Cys |
| Doria Lamba et al. (2007) | Family study | 7 | 71 | 1 | AD | Unknown | I | NH, SR, FI (6), AS (4), DM, NT, UH | p.Lys276Glu | p.Lys304Glu |
| Becker et al. (2006) | Family study | 7 | 83 | 6‡ | AR | Turkish | I | NH, SR, FI (1), AS (2), DM (1), NT (1) | deletion of exons 1-7 | deletion of exons 1-8 |
| Sirén et al. (2006) | Family study | 5 | 20 | 1‡ | AR | Turkish | I | NH (3), SR (1), Rg (1), FI (3), AS, DD (1), NT (1) | deletion of exons 1-7 | deletion of exons 1-8 |
| Poon et al. (2006) | Case report/series | 1 | 0 | 1 | AD | Asian | I | NH, SR | p.Tyr279Ser:het | p.Tyr307Ser:het |
| Coto et al. (2005) | Family study | 3 | 67 | 1 | AR | Caucasian | I | NH, SR | p.Arg72His | p.Arg100His |
| Tsai et al. (2004) | Family study | 2 | 0 | 1 | AR | Asian | I (1), U (1) | SR, Rg, FI, DD (1), NT(1) | p.Trp68Cys/p.Arg316X | p.Trp96Cys/p.Arg344X |
| Tijssen et al. (2003) | Original articles | 6 | 67 | 6 | AD | Caucasian | I | SR, FI, NT, CF (2) | p.Arg271Gln (4) | p.Arg299Gln |
| p.Lys276Glu (2) | p.Lys304Glu | |||||||||
| Miraglia Del Giudice et al. (2003) | Case report/series | 1 | 100 | 1 | AD | Caucasian | I | NH, SR, FI, AS, DD, DM, NT | p.Arg218Gln | p.Arg246Gln |
| Humeny et al. (2002) | Original articles | 1 | 100 | 1‡ | AR | Asian | I | SR, FI, DD, NT | p.Ser231Arg | p.Ser259Arg |
| del Giudice et al. (2001) | Case report/series | 1 | 100 | 1 | AD | Caucasian | I | NH, SR, Rg, NT | p.Val260Met | p.Val288Met |
| Kwok et al. (2001) | Case report/series | 3† | 33 | 2 | AD | Caucasian | I (2), U (1) | NH (2), SR, FI, DD (1), NT (2) | p.Arg271Gln (1) | p.Arg299Gln |
| p.Tyr279Cys (2) | p.Tyr307Cys | |||||||||
| Jungbluth et al. (2000) | Case report/series | 1 | 100 | AD | Caucasian | I | NH, SR, Rg, FI, DD, NT | p.Gly342Ser | p.Gly342Ser | |
| Vergouwe et al. (1999) | Family study | 2 | 50 | 1 | AR | Caucasian | I | NH, SR, FI, DD (1), NT, UH | p.Arg252His | p.Arg280His |
| Brune et al. (1996) | Original articles | 1 | 0 | 1‡ | AR | Turkish | I | NH, SR, AS, DD | Deletion of exons 1-6 | Deletion of exon 1-7 |
| Milani et al. (1996) | Family study | 4 | 25 | 1 | AD | Caucasian | I (1), U (3) | NH (2), SR (2), AS (1) | p.Gln266His | p.Gln294His |
| Rees et al. (1994) | Case report/series | 10 | 100 | 2 | AD | Caucasian | I (9), U (1) | NH, SR, Rg, FI, DM, UH (most) | p.Arg271Gln | p.Arg299Gln |
| p.lle244Asp | p.lle272Asp | |||||||||
| Ryan et al. (1992) | Family study | 29 | 41 | 1 | AD | unknown | I | NH, AS (5), FI (25), FD (1), DD (1), NT(1), IH (1) | p.Arg271leu | p.Arg299leu |
| Hayashi et al. (1991) | Family study | 9 | 67 | 2 | AD | Asian | I (7), U (2) | NH (5), SR (6), AS (4), FI (8), NT (7), IH (2), HD (2) | p.Arg271Gln | p.Arg299Gln |
| Kurczynski (1983) | Family study | 9 | 56 | 1 | AD | Caucasian | I | NH (9), SR (8), AS (1), FI (8), NT (3), UH (1) | p.Arg271Gln | p.Arg299Gln |
| Morley et al. (1982) | Family study | 15 | 33 | 1 | AD | Unknown | I | NH (12), SR, FI, NT (2) IH (3), HD (6) | p.Tyr279Cys | p.Tyr307Cys |
References of individual studies are summarized in Supplementary Material.
* who had mutation in GLRA1 gene,
† whose clinical data was descripted in detail,
‡ consanguineous marriage.
AD: autosomal dominant, AR: autosomal recessive, I: infant, C: childhood, U: unknown, NH: neonatal hypertonia, SR: exaggerated startle response, Rg: rigidity, Fl: falling attack, AS: apnea spells, FD: feeding difficulty, DD: developmental delay, DM: diurnal myoclonus, NT: nose tapping test, UH: umbilical hernia, HD: hip dislocation, IH: inguinal hernia, CF: club foot.
- 1. Zhou L, Chillag KL, Nigro MA. Hyperekplexia: a treatable neurogenetic disease. Brain Dev 2002;24:669–674.ArticlePubMed
- 2. Bakker MJ, van Dijk JG, van den Maagdenberg AM, Tijssen MA. Startle syndromes. Lancet Neurol 2006;5:513–524.ArticlePubMed
- 3. Giacoia GP, Ryan SG. Hyperekplexia associated with apnea and sudden infant death syndrome. Arch Pediatr Adolesc Med 1994;148:540–543.ArticlePubMed
- 4. Thomas RH, Chung SK, Wood SE, Cushion TD, Drew CJ, Hammond CL, et al. Genotype-phenotype correlations in hyperekplexia: apnoeas, learning difficulties and speech delay. Brain 2013;136(Pt 10):3085–3095.ArticlePubMedPDF
- 5. Tijssen MAJ, Rees MI. Hyperekplexia. In: Pagon RA, Adam MP, Ardinger HH, Wallace A, Bean LJH, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993-2016.
- 6. Shiang R, Ryan SG, Zhu YZ, Hahn AF, O’Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet 1993;5:351–358.ArticlePubMedPDF
- 7. Thomas RH, Drew CJ, Wood SE, Hammond CL, Chung SK, Rees MI. Ethnicity can predict GLRA1 genotypes in hyperekplexia. J Neurol Neurosurg Psychiatry 2015;86:341–343.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Hereditary hyperekplexia: a new family and a systematic review of GLRA1 gene-related phenotypes
Elisabetta Ferraroli, Marco Perulli, Chiara Veredice, Ilaria Contaldo, Michela Quintiliani, Martina Ricci, Ilaria Venezia, Luigi Citrigno, Antonio Qualtieri, Patrizia Spadafora, Francesca Cavalcanti, Domenica Immacolata Battaglia
Pediatric Neurology.2022;[Epub] CrossRef - Paroxysmal Nonepileptic Events in Children
Ilaria Lagorio, Lorenzo Brunelli, Pasquale Striano
Neurology Clinical Practice.2022; 12(4): 320. CrossRef - Four Turkish families with hyperekplexia: A missense mutation and the exon 1–7 deletion in the GLRA1 gene
Didem Tezen, Gülşah Şimşir, Özlem Çokar, Veysi Demirbilek, A. Nazlı Başak, Zuhal Yapıcı
Parkinsonism & Related Disorders.2022; 105: 128. CrossRef - Advances in hyperekplexia and other startle syndromes
Fei-xia Zhan, Shi-Ge Wang, Li Cao
Neurological Sciences.2021; 42(10): 4095. CrossRef - A Case of Hyperekplexia That Started From Childhood: Clinical Diagnosis With Negative Genetic Investigations
Annibale Antonioni, Giovanni Peschi, Enrico Granieri
Frontiers in Neurology.2020;[Epub] CrossRef - Excessive Startle with Novel GLRA1 Mutations in 4 Chinese Patients and a Literature Review of GLRA1-Related Hyperekplexia
Feixia Zhan, Chao Zhang, Shige Wang, Zeyu Zhu, Guang Chen, Mingliang Zhao, Li Cao
Journal of Clinical Neurology.2020; 16(2): 230. CrossRef - C.292G>A, a novel glycine receptor alpha 1 subunit gene (GLRA1) mutation found in a Chinese patient with hyperekplexia
Yan Zhang, Ling-Ling Wu, Xiao-Lan Zheng, Cai-Mei Lin
Medicine.2020; 99(17): e19968. CrossRef - Hyperekplexia and other startle syndromes
Arushi Gahlot Saini, Sanjay Pandey
Journal of the Neurological Sciences.2020; 416: 117051. CrossRef - Clinical features and genetic analysis of two siblings with startle disease in an Italian family: a case report
Teresa Sprovieri, Carmine Ungaro, Serena Sivo, Michela Quintiliani, Ilaria Contaldo, Chiara Veredice, Luigi Citrigno, Maria Muglia, Francesca Cavalcanti, Sebastiano Cavallaro, Eugenio Mercuri, Domenica Battaglia
BMC Medical Genetics.2019;[Epub] CrossRef - Weird Laughing in Hyperekplexia: A new phenotype associated with a novel mutation in the GLRA1 gene?
Zhi Huang, Yajun Lian, Hongliang Xu, Haifeng Zhang
Seizure.2018; 58: 6. CrossRef - A novel compound mutation in GLRA1 cause hyperekplexia in a Chinese boy- a case report and review of the literature
Zhiliang Yang, Guilian Sun, Fang Yao, Dongying Tao, Binlu Zhu
BMC Medical Genetics.2017;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite