 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 12(2); 2019 > Article
-
Letter to the editor
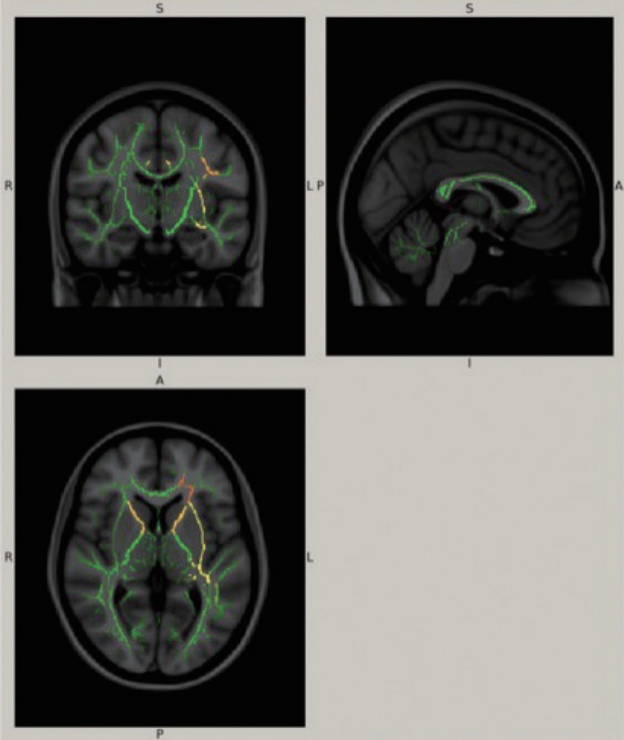
Alternating Hemiplegia of Childhood in a Person of Malay Ethnicity with Diffusion Tensor Imaging Abnormalities -
Ai Huey Tan1
 , Tien Lee Ong2, Norlisah Ramli3, Li Kuo Tan3, Jia Lun Lim4, Mohamad Addin Azhan1, Azlina Ahmad-Annuar4, Khairul Azmi Ibrahim2, Zariah Abdul-Aziz2, Laurie J. Ozelius5, Allison Brashear6, Shen-Yang Lim1
, Tien Lee Ong2, Norlisah Ramli3, Li Kuo Tan3, Jia Lun Lim4, Mohamad Addin Azhan1, Azlina Ahmad-Annuar4, Khairul Azmi Ibrahim2, Zariah Abdul-Aziz2, Laurie J. Ozelius5, Allison Brashear6, Shen-Yang Lim1 -
Journal of Movement Disorders 2019;12(2):132-134.
DOI: https://doi.org/10.14802/jmd.18063
Published online: May 30, 2019
1Division of Neurology and the Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, Department of Medicine, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
2Department of Medicine, Hospital Sultanah Nur Zahirah, Kuala Terengganu,Malaysia
3Department of Biomedical Imaging, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
4Department of Biomedical Science, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
5Departments of Genetics and Genomic Sciences and Neurology, Mount Sinai School of Medicine, New York, NY, USA
6Department of Neurology, Wake Forest University School of Medicine, Winston-Salem, NC, USA
- Corresponding author: Ai Huey Tan, MD, MRCP Neurology Laboratory, 6th Floor, South Tower, University of Malaya Medical Centre, 50603 Kuala Lumpur, Malaysia / Tel: +60-3-79492891 / Fax: +60-3-79492030 / E-mail: aihuey.tan@gmail.com
Copyright © 2019 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supplementary Materials
Supplementary Video Legends
Supplementary Table 1
- This work was supported by the Parkinson’s Disease and Movement Disorders Research Program, University of Malaya (PV035-2017); University of Malaya Research Grant (RP052B-17HTM); and NINDS R01NS058949 (AB).
- A written consent was obtained from the patient’s legal guardian prior to the publication of this case report. We would like to acknowledge the patient and her family for their participation in this case report.
Acknowledgments
- 1. Masoud M, Prange L, Wuchich J, Hunanyan A, Mikati MA. Diagnosis and treatment of alternating hemiplegia of childhood. Curr Treat Options Neurol 2017;19:8.ArticlePubMedPDF
- 2. Heinzen EL, Arzimanoglou A, Brashear A, Clapcote SJ, Gurrieri F, Goldstein DB, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol 2014;13:503–514.ArticlePubMedPMC
- 3. Wong YQ, Tan LK, Seow P, Tan MP, Abd Kadir KA, Vijayananthan A, et al. Microstructural integrity of white matter tracts amongst older fallers: a DTI study. PLoS ONE 2017;12:e0179895. ArticlePubMedPMC
- 4. Mori S, Oishi K, Jiang H, Jiang L, Li X, Akhter K, et al. Stereotaxic white matter atlas based on diffusion tensor imaging in an ICBM template. Neuroimage 2008;40:570–582.ArticlePubMedPMC
- 5. Erro R, Bhatia KP. Unravelling of the paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry 2019;90:227–234.ArticlePubMed
- 6. Panagiotakaki E, De Grandis E, Stagnaro M, Heinzen EL, Fons C, Sisodiya S, et al. Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood: a study of 155 patients. Orphanet J Rare Dis 2015;10:123.ArticlePubMedPMC
- 7. Sasaki M, Ishii A, Saito Y, Morisada N, Iijima K, Takada S, et al. Genotype–phenotype correlations in alternating hemiplegia of childhood. Neurology 2014;82:482–490.ArticlePubMed
- 8. Sampson JB, Michaeli TH, Wright BA, Goldman JE, Vonsattel JP, Fahn S. Basal ganglia gliosis in a case of rapid-onset dystonia-parkinsonism (DYT12) with a novel mutation in ATPase 1A3 (ATP1A3). Mov Disord Clin Pract 2016;3:618–620.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- White matter and cerebellar involvement in alternating hemiplegia of childhood
Mariasavina Severino, Livia Pisciotta, Domenico Tortora, Benedetta Toselli, Michela Stagnaro, Ramona Cordani, Giovanni Morana, Anna Zicca, Svetlana Kotzeva, Clelia Zanaboni, Giovanni Montobbio, Andrea Rossi, Elisa De Grandis
Journal of Neurology.2020; 267(5): 1300. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
